


| Publication number | WO2012168364 A1 |
| Publication type | Application |
| Application number | PCT/EP2012/060800 |
| Publication date | Dec 13, 2012 |
| Filing date | Jun 7, 2012 |
| Priority date | Jun 10, 2011 |
| Also published as | EP2718262A1, US8884016, US20140058107,US20150025242 |
| Inventors | Chiara Vladiskovic, Emanuele Attolino,Alessandro Lombardo, Simone TAMBINI |
| Applicant | Dipharma Francis S.R.L. |
| External Links: Patentscope, Espacenet | |
http://www.google.com/patents/WO2012168364A1?cl=en
l-(4-Methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l -yl)phenyl]-4, 5,6,7- tetrahydro- lH-pyrazolo[3,4-c]pyridine-3-carboxyamide of formula (I), also known come apixaban, is a powerful inhibitor of coagulation factor Xa disclosed in US 6,967,208. Said compound is used in the prevention and treatment of thromboembolic disorders.

(I)
US 7, 153,960 discloses a process for the preparation of apixaban wherein the key step is the formation of intermediate (A) by 1 ,3 dipolar cycloaddition reaction between the compounds of formula (B) and (C) and its subsequent conversion to the compound of formula (D) by treatment with an acid. The compound of formula (D), after simple manipulations of functional groups, is converted to apixaban

B C A D
Said patent discloses the preparation of the compounds of formula (B) and (C). While the synthesis of the hydrazone of formula (B) has been known for some time, the preparation of the key intermediate of formula (C) is complex and uses reagents which are expensive and potentially hazardous, such as phosphorus pentachloride (PC15), and drastic reaction conditions.
US 7, 153,960, for example, discloses as preferred the preparation of an enamine intermediate of formula (C) wherein the amine residue NRbRc is a morpholine. The conditions used for the success of the reaction actually involve the use of morpholine as solvent at high temperatures, such as reflux temperature (about 130- 135°C).
The complexity of the known processes for the preparation of the intermediate of formula C, the expense and danger of the reagents and the drastic reaction conditions used make said processes difficult to apply and scale up industrially, especially for the purpose of preparing the intermediates of formula A and D and apixaban.
There is consequently a need for an alternative method for the preparation of apixaban and its intermediates which does not involves the problems described above. Said method should in particular be more industrially scalable, allow the desired compounds to be obtained with high yields, and use cheaper reagents which are simpler to handle, also using mild reaction conditions.

Example 6. Synthesis of compound of formula (I): l-(4- Methoxyphenyl)-6-[4-(2-oxo-piperidinyl)phenyl]-7-oxo-4,5,6,7-tetrahydro- l//-pyrazolo[3,4-c]pyridine-3-carboxyamide: Apixaban (I)
The compound of formula II, prepared as in Example 5 (17.50 g, 35.82 mmol), is suspended in 100 ml of 33% NH3 and 200 ml of MeOH in a 1L 4-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere, and heated to 45°. MeOH (250 ml) is added until completely dissolved, and the solution is left under stirring for 2h. Another addition of 33% NH3 (50 ml) is performed, and the progress of the reaction is monitored by TLC (AcOEt/MeOH 9: 1) and HPLC. After 18h the solvent is evaporated under low pressure, and the solid residue obtained is suspended in 200 ml of H2O and left under stirring for 2h. The white solid is filtered through a Buchner funnel, and washed with H2O (50 ml). The product of formula (I) is stove-dried at 50°C to a constant weight (12.60 g, yield 76%). The HPLC purity of the product exceeds 99%.
1H NMR (300 MHz, CDC13): 67.47 (2H, dd, J0=8.7 Hz, Ar-H), 7.31
(2H, dd, J0=8.7 Hz, Ar-H), 7.23 (2H, dd, J0=8.7 Hz, Ar-H), 6.93 (2H, dd, J0=8.7 Hz, Ar-H), 6.83 (1H, s, N-H), 5.53 (1H, s, N-H), 4.1 1 (2H, t, J=6.6 Hz, CH2CH2N), 3.81 (3H, s, Ar-OCH3), 3.59 (2H, m, NCH2CH2CH2CH2CO) 3.37 (2H, t, J=6.6 Hz, CH2CH2N), 2.55 (2H, m, NCH2CH2CH2CH2CO), 1.93 (4H, m, NCH2CH^CH2CH2CO).

Preparation of intermediates




 4.09 (2H, t, J=6.6 Hz, CH2(¾N), 3.81 (3H, s, Ar-OCH3), 3.32 (2H, t, J=6.6 Hz, (¾CH2N), 1.43 (3H, t, J=7.3 Hz, COOCH2CH3).
4.09 (2H, t, J=6.6 Hz, CH2(¾N), 3.81 (3H, s, Ar-OCH3), 3.32 (2H, t, J=6.6 Hz, (¾CH2N), 1.43 (3H, t, J=7.3 Hz, COOCH2CH3).


Preparation of intermediates
Example 1. Synthesis of a compound of formula (III): l-(4-Iodophenyl)-3-trimeth lsilyloxy-5,6-dihydro-l -pyridin-2-one (Ilia)
v in
The compound of formula V (2.00 g, 6.35 mmol) is suspended in 15 ml of toluene in a 50 ml 3-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere. Triethylamine (1.15 ml, 8.26 mmol) is added, and the mixture is cooled to 0°C. Chlorotrimethylsilane (0.97 ml, 7.64 mmol) is added drop by drop at a temperature of under 5°C. After the addition, the temperature is increased to 50°C, and after 5h the reaction mixture is cooled to room temperature, diluted with toluene (30 ml) and washed with H2O (1x10 ml) and a saturated solution of NaCl (1x10 ml). The organic phase is anhydrified on (Na2SO4), filtered and evaporated under low pressure. Compound (III) is obtained as a brown solid (2.23 g, yield 90%), which is used for the next stage without further purification.
1H NMR (300 MHz, CDC13): 6 7.67 (2H, dd, J=8.1 Hz, Ar-H), 7.06 (2H, dd, J=8.1 Hz, Ar-H), 5.81 (1H, t, J=4.8 Hz, CHCOSiMe3), 3.77 (2H, t, J=6.9 Hz, N-CH2), 2.48 (2H, m, ΟΗ2¾ϋΗ), 0.23 (9H, s, CHCOSi(CH3 ).

Dipharma Francis S.r.l.. Via Bissone, 5 20021 Baranzate (Milano) Italy Telephone +39 02 38 228.370. Fax +39 02 38228246. E-mail exclusives@dipharma.it
Dipharma Francis S.r.l.. Via Bissone, 5 20021 Baranzate (Milano) Italy Telephone +39 02 38 228.370. Fax +39 02 38228246. E-mail exclusives@dipharma.it
Example 2. Synthesis of a compound of formula (III): l-(4-
V III
The compound of formula V (370 mg, 1.17 mmol) is suspended in 2.5 ml of toluene in a 25 ml 3-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere. Triethylamine (0.250 ml, 1.80 mmol), dimethylaminopyridine (16 mg, 0.132 mmol) and tosyl chloride (240 mg 1.26 mmol) are then added in sequence. The reaction proceeds at room temperature; after 16h the reaction mixture is diluted with 25 ml of AcOEt and washed with H2O (2x10 ml) and a saturated solution of NaCl (1x10 ml). The organic phase is anhydrified with Na2SO4, filtered and evaporated under low pressure. The compound of formula (III) is obtained as a brown solid (470 mg, yield 87%), which is used for the next stage without further purification.
1H NMR (300 MHz, CDC13): 67.92 (2H, dd, J0=8.4 Hz, Ar-H), 7.66 (2H, dd, Jo=9.0 Hz, Ar-H), 7.31 (2H, dd, J0=8.4 Hz, Ar-H), 6.96 (2H, dd, Jo=9.0 Hz, Ar-H), 6.66 (1H, t, J=4.5 Hz, CHCOTs), 3.82 (2H, t, J=6.9 Hz, N-CH2), 2.65 (2H, dt, J1=6.9 Hz, J2=4.5 Hz CH^HaCHOTs), 2.42 (3H, s, Ar-CH3).

Example 3. Synthesis of a compound of formula (IV): Chloro[(4- methoxyphenyl)hydrazono] acetic acid ethyl ester (IV)
IV p-anisidine (31.02 g, 0.252 mol) in 100 ml of H2O is suspended in a 250 ml 4-necked flask equipped with coolant, thermometer, dropping funnel and magnetic stirrer, and cooled to 0°C in an ice bath. 60 ml of 37% HC1 is added, followed by a solution of NaNO2 (20.95 g, 0.307 mol) in 50 ml of H2O, through a dropping funnel, maintaining the temperature below 5°C. After the addition the mixture is left under stirring at between 0 and 5°C for lh, and the solution obtained is added at 0°C to a solution containing ethyl-2- chloroacetoacetate (41.65 g, 0.254 mol), AcONa (47.84 g, 0.583 mol), AcOEt (200 ml) and H2O (100 ml). The biphasic system is stirred at between 0 and 5°C for lh, and then at about 25 °C for 16h. The phases are then separated and the organic phase is washed with a saturated solution of NaHCO3 (4x80 ml), anhydrified on Na2SO4, filtered and evaporated under low pressure. The compound of formula (IV) is obtained as a solid (about 57 g), which is used for the next stage without further purification.
1H NMR (300 MHz, CDC13): 68.24 (1H, s, N-H), 7.16 (2H, dd, Jo=9.0
Hz, Ar-H),6.88 (2H, dd, Jo=9.0 Hz, Ar-H), 4.37 (2H, q, J=7.2 Hz, COOCH2CH3), 3.79 (3H, s, Ar-OCH3), 1.39 (3H, t, J=7.2 Hz, COOCH2CH3).
Example 4. Synthesis of a compound of formula (II): l-(4- methoxyphenyl)-6-(4-iodophenyl)-7-oxo-4,5,6,7-tetrahydro-l//- pyrazolo[3,4-c]pyridine-3-carboxylic acid ethyl ester
III IV II
Compound (IV) (0.82 g, 3.19 mmol) is dissolved in 5 ml of AcOEt in a ml 3-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere, and cooled to 0° with an ice bath. Compound (III) (1.02 g, 2.63 mmol) and triethylamine (0.89 ml, 6.40 mmol) are added. When the additions have been completed, the reaction is heated to reflux temperature for 3h, after which another portion of compound (IV) (350 mg) and triethylamine (0.25 ml) are added. When compound (III) has completely disappeared, the end-of-reaction mixture is cooled to 0°C, treated with a solution of HC1 (1.80 g of 37% HC1 in 3.20 g of H2O) and maintained under stirring at room temperature overnight. A solid precipitates, which is filtered through a Buchner funnel and washed with 5 ml of a 1 : 1 solution of AcOEt and isopropanol. The compound of formula (II) is obtained as a solid (0.85 g, yield 63%).
1H NMR (300 MHz, CDC13): 67.67 (2H, dd, J0=8.7 Hz, Ar-H), 7.46 (2H, dd, Jo=9.0 Hz, Ar-H), 7.06 (2H, dd, Jo=9.0 Hz, Ar-H), 6.90 (2H, dd, Jo=9.0 Hz, Ar-H), 4.45 (2H, q, J=7.2 Hz,
Example 5. Synthesis of a compound of formula (II): l-(4- methoxyphenyl)-6-[4-(2-oxo-piperidinyl)phenyl]-7-oxo-4,5,6,7-tetrahydro- l//-pyrazolo[3,4-c]pyridine-3-carboxylic acid ethyl ester
II II
Compound II, prepared as in Example 4 (35.90 g, 69.40 mmol) is suspended in 250 ml of toluene in a 1L 4-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere, δ-valerolactam (13.74 g, 138.60 mmol), K3PO4 (30.25 g, 142.50 mmol) and Cul (2.54 g, 13.34 mmol) are then added in sequence. The suspension obtained is degassed 3 times at room temperature; N,N'-dimethylethylenediamine (1.65 ml, 26.78 mmol) is then added and the mixture is heated to reflux temperature. After 48 h the end-of-reaction mixture is filtered through a Buchner funnel, and the filter is washed with 200 ml of toluene. The toluene phase is washed with a solution of Na2S2O3 (50 g in 160 ml of H2O, 2x80 ml), 15% NH3 (2x80 ml) and a saturated solution of NaCl (1x80 ml). The organic phase is anhydrified on (Na2SO4), filtered and evaporated under low pressure. A solid product is obtained (37 g), which is crystallised by AcOEt. After crystallisation the product is obtained as a pure white solid (22.7 g, yield 67%).
1H NMR (300 MHz, DMSO-dtf): 67.47 (2H, dd, J0=8.7 Hz, Ar-H), 7.32 (2H, dd, Jo=9.0 Hz, Ar-H), 7.28 (2H, dd, J0=8.7 Hz, Ar-H), 6.90 (2H, dd, Jo=9.0 Hz, Ar-H), 4.32 (2H, q, J=6.9 Hz, COOCEbCH^, 4.06 (2H, t, J=6.6 Hz, CH2CH2N), 3.79 (3H, s, Ar-OCH3), 3.57 (2H, m, N(¾CH2CH2CH2CO) 3.19 (2H, t, J=6.6 Hz, ¾CH2N), 2.36 (2H, m, NCH2CH2CH2CH2CO), 1.83 (4H, m, NCH2CH2CH2CH2CO), 1.31 (3H, t, J=6.9 Hz, COOCH2CH3).











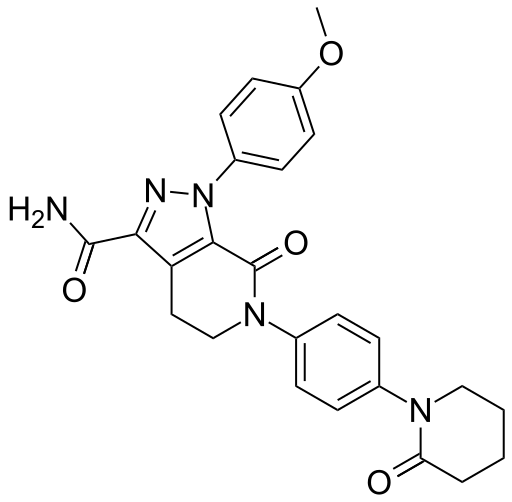 APIXABAN
APIXABAN

 .
.