
Process of reviewing and assessing the dossier to support a medicinal product in view of its marketing (also called licensing, registration, approval, etc.), obviously finalized by granting of a document also called marketing authorization (equivalent: product license). This process is performed within a legislative framework which defines the requirements necessary for application to the concerned (competent) regulatory authority, details on the assessment procedure (based on quality, efficacy and safety criteria) and the grounds for approval or rejection of the application, and also the circumstances where a marketing authorization already granted may be withdrawn, suspended or revoked.NOTE [1]
The application dossier for marketing authorization is called New Drug Application (NDA) in the USA or Marketing Authorization Application (MAA) in the European Union and other countries, or simply registration dossier. Basically, this consists of a dossier with data proving that the drug has quality, efficacy and safety properties suitable for the intended use, additional administrative documents, samples of finished product or related substances and reagents necessary to perform analyzes of finished product as described in that dossier. The content and format of the dossier must follow rules as defined by the competent authorities. For example, since year 2003, the authorities in the United States, the European Union and Japan ask for the Common Technical Document (CTD) format, and more recently, its electronic version - the electronic Common Technical Document (eCTD).

The application is filed with the competent drug regulatory authority in the concerned country, which can be either an independent regulatory body or a specialized department in the ministry of health.
In accordance with local legislation, the resulting document allowing to the applicant to market the product may be more detailed (in addition to data identifying the product and its holder it may contain addresses of all manufacturing sites, appended labeling, artwork of packaging components, etc.) until a one-page document called certificate of registration (and containing minimal data identifying the product and its source).
Many generic drugs are now being prescribed and the trend is increasing. For example, in Austria, the number of all generics prescriptions has more than doubled from 11% in 2000 to 23% in 2010. However, many myths and questions about generic drugs remain and information may be difficult to come by. It is therefore not surprising, as we have discovered in recent years, that even physicians and pharmacists are not always fully up to date in their understanding of generic drugs. Some of their questions centre on issues such as: are generic drugs really as good as the original; are we really dealing with an adequately tested, high quality medicinal product.
Today, generic drugs present an equally well-tolerated and efficacious alternative to established medicinal products, which contain well-known, rigorously tested active ingredients. An established originator product undergoes expensive and protracted development (up to 15 years) with inherently high preclinical and clinical research costs in order to be given market approval. The development of generic drugs, on the other hand, is relatively quick and inexpensive, which allows generic drugs to be sold at a distinctly cheaper price. This is due to the waiving of new preclinical and clinical studies, aside from some bioequivalence studies. Their lower price however should not be equated with ‘cheap quality’. In fact, generic medicines undergo the same strict scrutiny by the European or national medicines authorities as reference products.
This marketing authorisation validates the safety, efficacy, and quality of a generic drug.
What makes a generic medicinal product generic?
The definition, according to Austrian drug law/the Medicinal Products Act as well as to EU Directive 2001/83/EC, is that a generic medicinal product ‘is a product which has the same qualitative’, i.e. kind of active substance, ‘and quantitative’, i.e. amount of active substance, ‘composition as the reference medicinal product’. For the sake of simplicity, the reference product is often also referred to as the originator. The different salts, esters, or derivatives of an active substance are considered to be the same active substance, unless they differ significantly in their safety and/or efficacy properties. In these cases, the manufacturer of a generic drug has to submit further proof of efficacy and safety.
The pharmaceutical form, which means the distinct way a product is to be administered, of the generic medicinal product has to be the same as for the reference medicinal product [1]. However, remarkably the various types of immediate release oral pharmaceutical forms, e.g. tablets, capsules and dragées, are considered to be one and the same pharmaceutical form. A patient prescribed with a particular medicinal product may therefore be prescribed either a film-coated tablet or a capsule of the same drug by his physician. This in itself does not pose a problem, as the galenic formulation may indeed be different, but the impact on the safety and efficacy profile of the whole product has been judged to be comparable during the approval procedure.

Are different compositions possible?
Differences in composition between the generic and reference medicinal product are possible, but only regarding the excipients, e.g. bulking agents, colouring agents; and not for the active substances. For example, corn starch may be used instead of lactose as an excipient. However, it has to be demonstrated by the applicant of the generic drug that these differences in composition do not influence the therapeutic efficacy and safety or how the drug is absorbed, distributed, metabolised or eliminated by the body, i.e. the drug’s pharmacokinetics must also remain more or less the same.
Bioavailability or bioequivalence trials need to be conducted in order to demonstrate the equivalence between the generic medicinal product and the reference medicinal product. Differences in the manufacturing process compared to the originator are allowed, but the same strict general quality criteria, e.g. controlled production under good manufacturing practice (GMP), apply to the production of the generic medicinal product as well as for the reference product.
Also a medicinal product can only be considered as the reference product if it has been granted market approval in at least one Member State of the European Economic Area. However, only the so-called originator can serve as the reference product in bioequivalence testing, but never another generic drug, as this would otherwise mean the allowance of a copy of a copy.
How soon can generic medicinal products appear on the market?
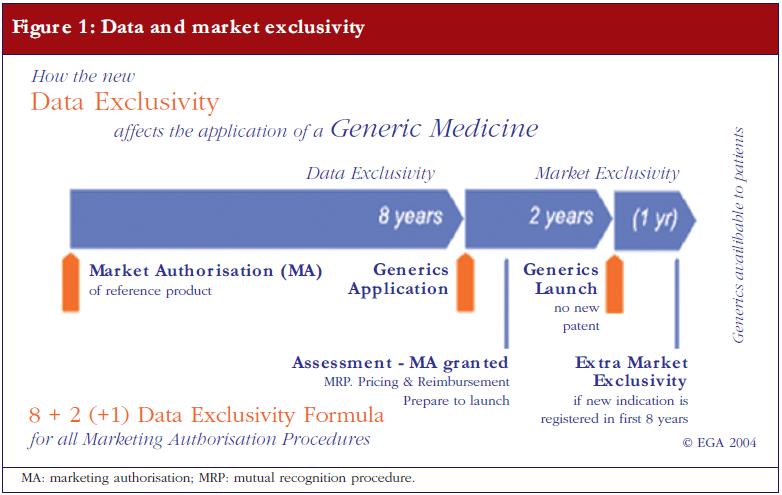
The applicant needs to provide proof that the originator product has been authorised for at least eight years, or that the originator company has issued a written informed consent stating that the generics company is permitted to apply for its generic drugs sooner. As a rule however, the earliest a generic medicinal product is allowed to go on sale is 10 years after the first European originator is granted marketing authorisation. This 10-year market exclusivity can be extended by an additional year if, during the first eight years, the marketing authorisation holder of the originator obtains an additional authorisation for one or more new relevant therapeutic indications [2]. Figure 1 shows the ‘8+2 (+1) Formula’ applicable to generic medicinal products entering the market.
Some originators, however, hold patents—in some cases up to 1,000 patents for one single product were found—which can further postpone the launch of a generic drug. Such delay in market access is therefore possible, even if the marketing authorisation has already been granted to the generic drug. Notably, some misuse of patent strategies was described in the final report of the 2009 sector inquiry of the European Commission for Competition [3]. In a sample of 219 molecules from 2000 to 2007 there were reportedly 1,300 patent-related out-of-court disputes related to the launch of a generics.
The number of patent litigations brought to court totalled nearly 700 cases in these seven years and the number of cases increased by a factor of four between 2000 and 2007. The report reached the conclusion that the behaviour and practices of originators contribute to generics delay as well as to difficulties in innovation itself because originators may even block each other.

What is a bioequivalence study?
Bioequivalence studies are often the demanded basis for granting marketing authorisation for a generic medicinal product. They are clinical studies conducted in accordance with Austrian drug law as well as to EU Directive 2001/20/EC and provide data to demonstrate bioequivalence between a test product, i.e. the generic medicinal product, and a reference product, i.e. the originator.
The rate and extent of absorption of the medicinal products and therefore the bioavailability of the active substance(s) are determined. It is a widely accepted regulatory assumption, even sometimes challenged by generics disputants, that equivalent plasma concentration time curves represent equivalent efficacy and safety. Therefore, if bioequivalence can be shown after the administration of the same molar dose, equivalence or assumption of so-called essential similarity of the two products in terms of efficacy and safety can be concluded.
What are the rules for conducting bioequivalence studies?
How exactly a bioequivalence study has to be conducted, and which requirements need to be taken into consideration, is laid out in detail in the European bioequivalence guideline, the revised version of which came into effect mid-2010. The guideline clearly specifies the requirements for the design, conduct, and evaluation of bioequivalence studies for all EU countries. Since 2001, when the first bioequivalence guideline was published, many additional aspects were identified which needed to be amended and improved. Minor issues were addressed in interim question and answer documents.
After a three-year preparation period, the comprehensively revised version of the guideline came into effect in August 2010. The comments and suggestions of over 50 expert organisations and associations were worked into the 22 draft versions. The revised version therefore now reflects the most up-to-date state of knowledge, which is essential in issuing harmonised and standardised marketing authorisations across Europe.
The aim of the guideline was to do away with the ambiguities of the past, which often led to lengthy discussions and differences in professional opinion between the countries and competent authorities of the EU. This also ensures the safety and efficacy of all generic drugs being granted marketing authorisation.
Are bioequivalence studies only used in the development of generic drugs?
Since the task of bioequivalence studies is to detect differences between formulations or pharmaceutical forms, they are indeed not only used as a basis for the licensing of generic medicinal products. In fact, originators may also use bioequivalence studies during their own development since the formulation first used in clinical trials is often not the same which later goes into large-scale market production. Bioequivalence studies are used in these cases to allow bridging of the results obtained in the clinical trials. The same principles in study conduct, data evaluation, and assessment of results by the authority are applied in such originator studies as in the above-described studies for generic drugs. Remarkably, essential similarity between an originator small-scale clinical trial product and a large-scale originator product later to be for sale has never been put in question by anyone. Considering media coverage sometimes casts massive doubt about generics and the way they are authorised, obviously there seems to be an unfounded contrast in the perception dependent on who—the originator company or the generics company—makes use of the bio – equivalence concept for the authorisation of one of their products. Assuming that the bioequivalence concept is valid and trustworthy for authorisation of a new originator product, the same should be applied to the authorisation of a generic drug.
Is the manufacturing quality the same for generic and originator products?
The same quality requirements apply to the manufacturing of generic drugs as for any other medicinal product. Production has to be performed in accordance with GMP and is strictly controlled by evaluating the manufacturing data and by inspections performed not only in Austria and the EU, but also all over the world including countries such as India and South Africa. As for any other medicinal product, quality deficiencies in individual batches are theoretically possible and therefore the Austrian Federal Office for Safety in Health Care as well as the other competent EU authorities closely monitor the quality of all authorised medicinal products on the market. This is achieved by the legal obligation of authorisation holders to inform the authority about every out-of-specification results or other problems in manufacturing and an additional quality-defect notification system involving all healthcare professionals. This guarantees that only high quality medicinal products are available, regardless of whether these products are originators or generics.
When is a generic drug granted market authorisation?
An Austrian or an EU marketing authorisation is only issued when the pharmacokinetic parameters of the generic drug are comparable to those of the reference product and bioequivalence has been successfully demonstrated.
Furthermore, the overall benefits of the generic medicine need to outweigh its risks (positive risk–benefit ratio) and its excipients and the manufacturing process must have been demonstrated to not negatively influence its safety and efficacy.
Last but not least, all internationally relevant quality standards and legal requirements have to be fulfilled before marketing authorisation can be granted.
Procedures for obtaining a marketing authorization
Authorization processes follow either a purely national procedure, with rules and requirements as per national legislation in force, as it occurs in most of countries worldwide, or should follow a centrally approval or a mutual recognition or decentralized procedure within the European Union.
Types of applications
The type of application may vary according to status of the active ingredient.
Thus, if the application concerns a new active ingredient (new active substance, new chemical entity, new molecular entity), one talks about a full application.
Once a new active ingredient authorized, any additional strengths, pharmaceutical forms, administration routes, presentations, as well as any variations (changes to the existing marketing authorization) and extensions shall also be granted an authorization or be included in the initial marketing authorization, being subject of an abridged application.NOTE [2]
Special consideration is to be given to application for authorization of biological products and biotechnology products,[1] homeopathic products, herbal drugs, radionuclide generators, kits, radionuclide precursor radiopharmaceuticals and industrially prepared radiopharmaceuticals; in such instances, requirements are specific, in the meaning that they are special, more or less detailed, as per the nature of active ingredient.
Validity of marketing authorizations
In most countries, a marketing authorization is valid for a period of 5 years. After this period, one should apply for renewal of the marketing authorization, usually by providing minimal data proving that quality, efficacy and safety characteristics are maintained and the risk-benefit ratio of the medicinal product is still favourable. However, in the European Union, after one renewal, the marketing authorization shall remain valid for an unlimited period, unless the competent regulatory authority decides otherwise.NOTE [3]
If the marketing authorization is not renewed in a due time as requested by the local legislation, in order to maintain the pharmaceutical product on a market, one can apply for re-authorization (re-registration). In such situations, the applicant may be requested to submit the whole items necessary for a full application.
Marketing authorization may be withdrawn, suspended, revoked or varied by regulatory authorities if under normal conditions of use the benefit over risk ratio is no more favorable, the product is harmful, or if it lacks therapeutic efficacy; also, one of the above actions can be taken if the qualitative and quantitative composition or other qualitative aspects (control) are not as currently declared.
Marketing authorization may be also withdrawn, suspended or revoked if the marketing authorization holder or its representative does not fulfill other legal or regulatory obligations necessary to maintaining of product on the market, as per the legislation in force.
Also, the marketing authorization is withdrawn in the EU if the product is not placed on the market within next 3 consecutive years after granting of authorization or if it is no more marketed for 3 consecutive years (so-called “sunset clause”).NOTE [4]
Notes
- ^ a b http://www.mhra.gov.uk/Howweregulate/Medicines/Licensingofmedicines/Marketingauthorisations/index.htm#3
- ^ Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. Official Journal of the European Union, L 311, 28.11.2001, p. 67.
- ^ Directive 2004/27/EC of the European Parliament and of the Council of 31 March 2004 amending Directive 2001/83/EC on the Community code relating to medicinal products for human use. L 136, 30.4.2004, p. 34.
- ^ CMD(h) Agreement on Sunset Clause and its application to MAs granted in more than one Member State. Co-ordination Group for Mutual Recognition and Decentralised Procedures -Human, December 2006.



