 ...
...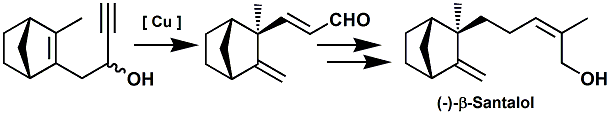

Charles Fehr and co-workers at
Firmenich have designed the "right cat for the desired
odor". In WIPO Patent Application
WO/2009/141781
(November 26, 2009), they disclose a novel, and seemingly
practical, approach to the synthesis of beta-Santalol (the
Golden Grail of Sandalwood odor).
See also - Charles Fehr,
Iris Magpantay, Jeremy Arpagaus, Xavier Marquet, Magali
Vuagnoux,
Enantioselective
Synthesis of (-)-beta-Santalol by a Copper-Catalyzed Enynol
Cyclization-Fragmentation Reaction, Angewandte Chemie,
Volume 121, Issue 39, Date: September 14, 2009, Pages:
7357-7359
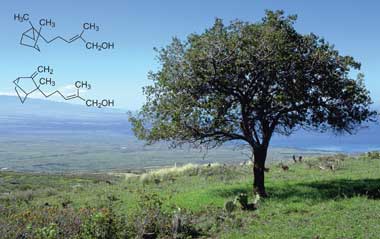
β-Santalol is an
organic compound that is classified as a
sesquiterpene. It comprises about 20% of the oil of
sandalwood, the major component being
α-santalol. As of 2002, about 60 tons of sandalwood oil are produced annually by
steam distillation of the heartwood of
Santalum album.
[1]
References
- Karl-Georg
Fahlbusch, Franz-Josef Hammerschmidt, Johannes Panten, Wilhelm
Pickenhagen, Dietmar Schatkowski, Kurt Bauer, Dorothea Garbe, Horst
Surburg "Flavors and Fragrances" in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim: 2002. Published online: 15 January 2003; doi:10.1002/14356007.a11_141.
Out of the woods
Woody
fragrances such as Iso E Super, a compound known as a 'floraliser' and
used in most new fine fragrances, also rate highly, says Gautier. Some
people smell a cedarwood note in Iso E Super, while others perceive it
to be musky. The male fragrance Fahrenheit (Dior, 1988) is 25 per cent Iso E Super while Perles de lalique
(Lalique, 2007) is 80 per cent Iso E Super. The commercial Iso E Super
owes its woody smell to a 5 per cent impurity which has an odour
threshold about 100 000 times lower than that of the main product.
Alpha- (top) and beta-santalol are constituents of the prized East Indian sandalwood oil
|
In
1999, Kraft's team at Givaudan isolated this impurity by epoxidation of
a commercial Iso E Super and proposed a structure based on NMR spectra.
The team came up with a way to synthesise the compound but it was not
commercially viable and the structure turned out to be too complex for
an alternative synthetic procedure. But out of several synthesised
analogues, one had an almost equal odour threshold and smelled even
better. What is more, its industrial synthesis is straightforward and it
has become a big hit - used by Givaudan as the captive material
Georgywood.
However, on strictly cash terms, the
most valuable fragrances are the natural products. For example, East
Indian sandalwood oil is one of the most precious and expensive
perfumery raw materials.
Beta-santalol is the main
olfactory constituent of sandalwood oil. It wasn't until 1990 that
beta-santalol could be prepared in the lab - this took an 11-step
reaction that was unsuitable for industrial scale-up. The best
sandalwood oil substitutes are derivatives of alpha-campholenic
aldehyde, prepared from inexpensive alpha-pinene, most of which is a
byproduct of the paper industry.
'The components in
sandalwood oil are not easy to make,' acknowledges Gautier. 'But what
Firmenich has done is screened all around that kind of structure to try
to find woody notes. We have a lot of substitutes so that perfumers can
create something close to sandalwood without needing sandalwood oil.'
Kraft agrees that there are 'excellent synthetic substitutes' out there.
For example, the campholenic aldehyde derivatives typically have a good
sandalwood smell. 'The other olfactory parts of natural sandalwood oil -
such as the smokey, cedar-like aspects - can easily be added using, for
example, cedarwood oil,' says Kraft.
Sandalwood was initially quite cheap (Guerlain's Samsara
contained 25 per cent sandalwood) but overharvesting has led to Indian
sandalwood trees being listed on the World Conservation Union's
threatened species red list.
'There's still a good
market for sandalwood oil and the price has gone, in the past six or
seven years, from $1000 [£680] per kg to over $2000,' says Leffingwell.
One Australian firm, TFS Corporation, has taken the initiative and
planted over 1700 hectares of the endangered Indian sandalwood (Santalum album ) in plantations in Western Australia.
http://www.google.com/patents/WO2009141781A1?cl=en
A) Preparation of β-santalol (via Wittig and hydroxyalkylation reaction)
A solution of butyllithium in hexanes (1.35 M, 11.7
ml, 15.8 mmol) was added over a 15 minutes period to a stirred
suspension of ethyltriphenylphosphonium iodide (6.61g, 15.8 mmol) in THF
(125 ml) at 0
0C. The resultant red solution was cooled to
-78°C and 3-(2-methyl-3-methylene-bicyclo[2.2.1]hept-2-exo-yl)-propanal
(2.55 g, 14.33 mmol) in solution in THF (16 ml) was added over a 15
minutes period. After further 5 minutes at -78°C, a solution of
butyllithium in hexanes (1.35 M, 12.7 mL, 17.2 mmol) was added over a 5
minutes period and the mixture was further stirred for 20 minutes at
-78°C before allowing to reach 0
0C in 2 hours. Dry
paraformaldehyde (2.60 g, 86.0 mmol) was added in one portion to the
deep red homogeneous solution and the mixture was stirred for 30 minutes
at 0
0C and was allowed to reach room temperature. After 1
hour at room temperature the mixture was poured into 5.2 ml of saturated
aqueous solution of NH
4Cl and extracted twice with CH
2Cl
2. The organic layer was washed with water and brine, and dried with Na
2SO
4.
The mixture was filtered through a short pad of silica gel with
dichloromethane as eluent and solvents were removed under pressure to
give a crude. Purification of crude compound was performed by flash
chromatography on silica gel with cyclohexane/AcOEt 90/10) as eluent to
give pure β-santalol as pale yellow oil. Further bulb to bulb
distillation under reduced pressure afforded β-santalol in 50% yield
(Z:E=95:5). 1H NMR (CDCl
3, 400 MHz): 5.29 (t, J=7.5, IH),
4.73 (s, IH), 4.45 (s, IH), 4.14 (s, 2H), 2.66 (d, J=3.8, IH), 2.12-1.94
(m, 3H), 1.78 (d, J=I.2, 3H), 1.71-1.60 (m, 3H),
1.44-1.36 (m, 2H), 1.32 (br s, IH), 1.27-1.17 (m,3H), 1.04 (s, 3H).
13C NMR (CDCl
3, 125 MHz): 166.2, 133.9, 129.0, 99.7, 61.6, 46.8, 44.7, 44.6, 41.5, 37.1, 29.7, 23.7, 23.2, 22.6, 21.2.
B) Preparation of β-santalol (via [1,4]
hydrogenation) i) Preparation of a compound (V):
2-methyl-5-(2-methyl-3-methylene-bicyclo[2.2.1]
hept-exo-2-yl)-pent-2-enal
3-(2-Methyl-3-methylene-bicyclo[2.2.1]hept-2-exo-yl)-propan-l-al (274.0 mg,
1.54 mmol) was dissolved in toluene (15.0 ml) at room
temperature under nitrogen. The mixture was heated to reflux and
propionaldehyde (0.4 ml, 1.96 mmol) and aqueous catalytic solution of
hexamethyleneimine and benzoic acid (0.12 ml, 0.616 mmol) was separately
added in one portion. Once that addition was finished, the mixture was
further heated at reflux for 6 hours. The mixture was cooled down to
room temperature and extracted twice with brine, the organic layer was
dried over MgSθ
2, filtered off and concentrated to give a
crude which was further purified by flash chromatography with
cyclohexane/ AcOEt (95/5) to afford the title compound in 80% yield.
1H NMR: 9.38 (s, IH), 6.48 (dt, J
1=?^, J
2=1.2,
IH), 4.78 (s, IH), 4.49 (s, IH), 2.69 (d, J=3.9, IH), 2.40-2.29 (m,
2H), 2.12 (d, J=3.1, IH), 1.75 (s, 3H), 1.72-1.64 (m, 3H), 1.59-1.51 (m,
IH), 1.47-1.36 (m, 2H), 130-1.21 (m, 2H), 1.09 (s, 3H).
13C NMR: 195.2, 165.5, 155.2, 139.1, 100.3, 46.8, 44.8, 44.7, 39.4, 37.1 , 29.6, 24.9, 23.7, 22.6, 9.1.
H) Preparation of a compound (IV): Acetic acid
2-methyl-5-(2-methyl-3-methylene-
bicyclo[2.2.1]hept-2-exo-yl)-penta-l,3-dienyl ester To a stirred
solution of 2-methyl-5-(2-methyl-3-methylene-bicyclo[2.2.1]hept-2-yl)-
pent-2-enal (268.0 mg, 1.23 mmol) in toluene (3.0 ml) were added Ac
2O (0.35 ml, 3.70 mmol), Et
3N
(0.70 ml, 5.02 mmol), and a catalytic amount of DMAP (15.0 mg, 0.1
mmol). The resulting mixture was heated to reflux for 22 hours. The
mixture was cooled down to room temperature and quenched with brine,
extracted twice with Et
2O, dried over MgSO
4,
filtered off and concentrated to give a crude which was further purified
by flash chromatography with cyclohexane/AcOEt (98/2) to afford the
title compound in 82% yield (E:Z=79:21).
1H NMR: 7.18 (s, IH), 5.99 (d, J=12.4, IH), 5.72 (dt, i
l=\2A, J
2=6.1,
IH), 4.76 (s, IH), 4.47 (s, IH), 2.68 (d, 3.4, IH), 2.17 (s, 3H),
2.12-2.01 (m, 2H), 1.81 (d, J=I.0, 3H), 1.73-1.63 (m, 3H), 1.43-1.39 (m,
2H), 1.27-1.18 (m, 2H), 1.02 (s, 3H).
13C NMR: 167.9, 165.5, 134.4, 130.6, 126.9, 120.7, 100.0, 46.9, 45.3, 45.0, 44.5, 37.0, 29.7, 23.6, 23.0, 20.8, 10.4.
Hi) Preparation of(2Z)-Acetic acid 2-methyl-5-(2-methyl-3-methylene-bicyclo[2.2.1] hept-2-exo-yl)-pent-2-enyl ester (VII)
Acetic acid
2-methyl-5-(2-methyl-3-methylene-bicyclo[2.2.1]hept-2-exo-yl)-penta-l,3-
dienyl ester (6.80 g, 93% pure; 24.3 mmol0.18 mmol) was treated with
[(Cp*)Ru(COD)]BF
4 (52 mg, 0.122 mmol) and maleic acid (230 mg, 1.95 mmol) in dry and degassed acetone (20 ml) at 60
0C under 4 bars of H
2 for 24 hours. The product was extracted with pentane/5% NaOH, washed twice with saturated aqueous NaCl, dried
(Na
2SO
4) and bulb-to-bulb
distilled: 6.80 g (81% Z and 5% E by GC; 92%). 1H NMR: 5.38 (t, J=7.1,
IH), 4.73 (s, IH), 4.59 (s, IH), 4.45 (s, IH), 2.66 (br s, IH),
2.12-2.04 (m, 4H), 2.07 (s, 3H), 1.73 (d, J= 1.0, 3H), 1.69-1.61 (m, 3H), 1.45-
1.37 (m, 2H), 1.27-1.17 (m, 3H), 1.04 (m, 3H).
13C NMR: 171.1, 166.1, 131.4, 129.4, 99.7, 63.2, 46.8, 44.7, 44.6, 41.2, 37.1, 29.7, 23.7,
23.4, 22.6, 21.5, 21.0.


................
see
http://pubs.rsc.org/en/content/articlelanding/2014/ob/c3ob42174k#!divAbstract
Biocatalyst mediated regio- and stereo-selective hydroxylation and epoxidation on (
Z)-α-santalol were achieved for the first time, using a fungal strain
Mucor piriformis. Four novel metabolites were characterized as 10,11-
cis-β-epoxy-α-santalol, 5α-hydroxy-(
Z)-α-santalol, 10,11-dihydroxy-α-santalol and 5α-hydroxy-10,11-
cis-β-epoxy-α-santalol. Using Amano PS lipase from
Burkholderia cepacia, α- and β-isomers of 10,11-
cis-epoxy-α-santalol were resolved efficiently.















.png)
