WO 2015155704, An improved process for the preparation of pramipexole dihydrochloride monohydrate
PIRAMAL ENTERPRISES LIMITED [IN/IN]; Piramal Tower, Ganpatrao Kadam Marg Lower Parel Mumbai 400013 (IN)
|
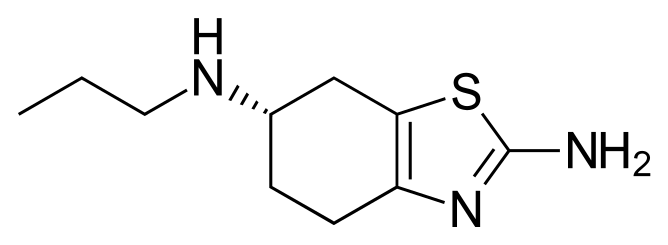
Pramipexole, (S)-2-amino-4,5,6,7-tetrahydro-6-(propylamino)benzothiazole, represented by the following formula I (the compound of formula I), is a dopamine D2/D3 agonist used for treatment of Schizophrenia, and particularly for the treatment of Parkinson's disease. Pramipexole is marketed in the form of dihydrochloride monohydrate salt under the brand name Mirapex.

Formula I
The compound of formula I is disclosed in US Patent no. 4,886,812 (US '812 Patent). The US' 812 Patent also describes a process for the preparation of the compound of formula I and its dihydrochloride monohydrate salt involving the propylation reaction of the compound of formula II with n-propylbromide as a propylating agent in the presence of potassium carbonate by using methanol as a solvent to provide the reaction mixture. The resulting reaction mixture is then refluxed for 3 hours. After completion of the reaction, water is added to the reaction mixture. The reaction mixture is then extracted with ethyl acetate and concentrated to obtain the residue. The obtained residue is purified by silica gel chromatography and the corresponding fraction is concentrated under reduced pressure to obtain the compound of formula I which is then converted into its dihydrochloride monohydrate salt. Although, US '812 Patent describes the process for the preparation of the compound of formula I from the compound of formula II, it does not teach the process for converting the compound of formula I into its dihydrochloride monohydrate salt. Also, the process described in US '812 Patent involves propylation of the compound of formula II using 4 molar equivalents of n-propylbromide as the propylating agent. N-propylbromide is known to be carcinogenic compound and its average threshold limit value for 8 hours exposure is 10 parts per million. Therefore, on commercial scale, excess use of such a hazardous reagent is not desirable. Further, propylation of the compound of formula II using the process described in US '812 Patent generates one major impurity namely (6S)-2,6-benzothiazolediamine,4,5,6,7-tetrahydro-N2,N6-dipropyl. The US '812 Patent does not teach any purification method for removal of this impurity.
Indian Patent Application no. 694/MUM/2006 describes a process for the preparation of the dihydrochloride monohydrate salt of the compound of formula I involving treating the alcoholic solution of the compound of formula I with hydrochloric acid and precipitating the dihydrochloride monohydrate salt of the compound of formula I by addition of water. The process disclosed in this patent application does not involve any purification step for the purification of the compound of formula I or its dihydrochloride monohydrate salt and thus, the final active pharmaceutical ingredient (API), the dihydrochloride monohydrate salt of the compound of formula I prepared by this process does not have the desired pharmaceutically acceptable purity.
Indian patent application no. 605/MUM/2008 describes a process for the preparation of the dihydrochloride salt of the compound of formula I. The process for the preparation of the dihydrochloride salt of the compound of formula I involves the propylation reaction of the compound of formula II with n-propanal as a propylating agent by using a mixture of methanol and water as the solvent. To the resulting reaction mixture, glacial acetic acid and sodium borohydride are charged and the reaction mixture is stirred for 30-40 minutes at a temperature of 15 to 20°C. The reaction mixture is then cooled to -5 to 0°C and to the reaction mixture; second lot of n-propanal with methanol and sodium borohydride is added. The resulting reaction mixture is stirred for 30-40 minutes and quenched with brine solution. The reaction mixture is distilled under vacuum to obtain a residue. To the obtained residue, ethyl acetate and water are added. Two layers formed are separated and ethyl acetate layer is concentrated under vacuum to obtain the crude compound of formula I. The resulting crude compound of formula I is then recrystallised by using acetonitrile to yield the pure compound of formula I. To the pure compound of formula I; ethanolic hydrochloric acid solution is added. The reaction mixture is stirred for 1 hour to precipitate the solid. The precipitated solid is filtered and suspended in ethanol. The reaction mixture is then stirred at reflux temperature for 30 minutes and at room temperature for 1 hour to precipitate the dihydrochloride salt of the compound of formula I. The precipitated dihydrochloride salt of the compound of formula I is dissolved in a mixture of ethanol and water; and filtered through hyflo. The filtrate is then distilled under vacuum and recrystallised by using ethanol to obtain the pure dihydrochloride salt of the compound of formula I. The process disclosed in said patent involves the use of 3 molar equivalents of sodium borohydride and n-propanal which renders the process costlier and hence, this process is not viable for scale up.
The general process for producing the dihydrochloride monohydrate salt of the compound of formula I is depicted in the following Scheme I:

(S)-2-amino-6-propinamido-4, 5,6,7- tetrahydrobenzothiazole
sodium borohydride

o e compoun o ormu a
Scheme I
Scheme-II.

methanol-water purification

Scheme-II
Examples
Example 1:
Step A: Synthesis of compound of formula I:
To the reaction flask dichloromethane (1500 ml), methanol (1500 ml) and the compound of formula II (100 gm) were charged at a temperature of 25-30° C. The reaction mixture was cooled to a temperature of 3-8 °C and to the reaction mixture, sulphuric acid (8.69 gm); n-propanal (13.98 ml) and sodium borohydride (2.46 g) were charged. The reaction mixture was stirred for 20-30 minutes at a temperature of 3-8°C. To the reaction mixture, n-propanal (41.94g) followed by sodium borohydride (7.38g) were added in three different lots at a temperature of 3-8°C. After completion of the reaction, the reaction mixture was quenched with brine solution. The quenched reaction mixture was further concentrated up to 15-16 volumes at 50-55°C under vacuum. The reaction mixture was cooled to 15-20°C. To the reaction mixture potassium carbonate (150 g), ethyl acetate (900 ml) and methanol (100 ml) were charged. The two layers formed were separated. The organic layer was then concentrated up to 7 to 8 volumes. To the organic layer ethyl acetate (500 ml) and brine solution (240 g) were added. The two layers formed were separated. The organic layer was treated with activated charcoal and filtered through hyflo. The organic layer was then concentrated under vacuum to obtain residue. To the obtained residue diisopropyl ether (200 ml) was added and reaction mixture was stirred for 20-30 minutes at 45-50°C. The reaction mixture was then cooled at 25-30°C to precipitate solid. The precipitated solid was then filtered and washed with diisopropyl ether (200ml) to obtain the compound of formula I.
Step B: Synthesis of monohydrochlonde salt of the compound of formula I:
To the reaction flask, the compound of formula I (as obtained in the step A) and isopropyl alcohol (900 ml) were charged and the reaction mixture was stirred at a temperature of 25-35°C for 1 hour. The reaction mixture was then filtered through hyflo and washed with isopropyl alcohol (100 ml). To the filtrate cone, hydrochloric acid (42.20 ml) was added to obtain a solid. The obtained solid was then filtered and washed with isopropyl alcohol (200 ml) to yield the monohydrochloride salt of the compound of formula I.
Step C: Purification of the monohydrochloride salt of the compound of formula I:
To the reaction flask, the monohydrochloride salt of the compound of formula I (as obtained in the step B) and the mixture of methanol (300 ml) and water (5.01 ml) were charged and the reaction mixture was stirred at a temperature of 55-60°C for 2 hours. The resulting reaction mixture was then cooled to a temperature of 20-25°C to precipitate solid. The precipitated solid was then filtered and washed with isopropyl alcohol (200 ml) to obtain the pure monohydrochloride salt of the compound of formula I.
Step D: Synthesis of the dihydrochloride monohydrate salt of the compound of formula I:
To the reaction flask, the pure monohydrochloride salt of the compound of formula I (as obtained in the step C), methanol (600 ml) and cone, hydrochloric acid (33.67 ml) were charged and the reaction mass was stirred at a temperature of 3-8°C for 2 hours. To the reaction mass, activated charcoal (4g) was charged and the reaction mass was stirred for 30-45 minutes at temperature of 40-50°C. The activated charcoal was filtered through hyflo and filtrate was concentrated under vacuum to obtain residue. To the residue, isopropyl alcohol (700 ml) was charged and the reaction mass was maintained for 2-3 hours at 15-20°C to precipitate solid. The precipitated solid was then filtered and washed with isopropyl alcohol (100 ml). The solid was then dried under vacuum to yield dihydrochloride monohydrate salt of the compound of formula I. Yield 36%, purity 99.77%.
Details for HPLC analysis:
Column: Inertsil ODS-3, 125 X 4.0 mm, 5μιη
Part No: C/N 5020
Mobile phase
Mobile phase A: Buffer solution
Mobile phase B: Acetonitrile: Buffer (500:500 v/v)
Flow rate: 1.5 ml/min
Injection volume: 5 μΐ
Run time: 25 minutes
Detector: 264 nm.
Column temperature: 40°C
Diluent: Acetonitrile: Buffer (200:800 v/v)
Procedure:
For system suitability inject (5μί) of the system suitability solution. The resolution between Pramipexole (the compound of formula I) related compound and Pramipexole should not be less than 6.0. The tailing factor for Pramipexole should not be more than 2.0. Inject Standard solution in six replicates into the chromatograph. For the Pramipexole peak, the relative standard deviation should not be more than 5.0%.
Inject (5μί) of blank preparation and test solution into the chromatograph, measure the responses of all the peaks and calculate all known impurities and unknown impurities by the formula given below. In the sample chromatogram disregard any peak due to the blank. Retention time and relative retention times are given in the table below.
Calculation :- SPL (Area) Cone STD
% impurities = X X 100
STD (Area) Cone SPL
Where:
SPL (Area) - is area of peak due to impurities in sample preparation.
STD (Area) - is mean area of peak of Pramipexole in reference solution (a) for injections.
Cone SPL - concentration of Pramipexole in test solution in mg/mL
Cone STD - concentration of Pramipexole in test solution in mg/mL

 Chairman of Piramal Enterprises Ltd. Ajay Piramal
Chairman of Piramal Enterprises Ltd. Ajay Piramal
Swati Piramal



















