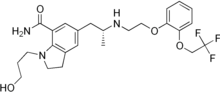Was developed and launched by Aegerion, under license from the University of Pennsylvania (which acquired rights from BMS).

Sanjay Jagdish DESAI
Brij KHERA
Jagdish Maganlal PATEL
Harshita Bharatkumar SHAH
Arunkumar Shyam Narayan UPADHYAY
Sureshkumar Narbheram AGRAVAT
Brij KHERA
Jagdish Maganlal PATEL
Harshita Bharatkumar SHAH
Arunkumar Shyam Narayan UPADHYAY
Sureshkumar Narbheram AGRAVAT
Polymorphic forms of lomitapide and its salts and processes for their preparation
Zydus Cadila Healthcare Ltd
The present invention relates to various polymorphic forms of lomitapide or its salts and processes for preparation thereof. The present invention provides Lomitapide mesylate in solid amorphous form and process for preparation thereof. The invention also provides an amorphous solid dispersion of lomitapide mesylate. Further, various crystalline forms of lomitapide mesylate like A, B and C and process for preparation thereof are provided. The invention also provides crystalline forms of lomitapide free base, in particular Form I and Form-II and their preparation. The invention further provides compositions comprising various forms of lomitapide and its salts.
A novel amorphous form of lomitapide mesylate (having >98% of purity and 0.5% of residual solvent and particles size D90 of >250 µm, D50 of >100 µm and D10 of >50 µm), a process for it preparation and a composition comprising it is claimed. Also claimed is an amorphous solid dispersion of lomitapide mesylate and a carrier (eg hydroxypropylmethyl cellulose acetate succinate). Further claimed are crystalline forms of lomitapide mesylate (designated ad Forms A, B, C, I, II and free base of lomitapide in amorphous form), processes for their preparation and compositions comprising them. Lomitapide is known to act as a microsomal triglyceride transfer protein inhibitor, useful for treating familial hypercholesterolemia.
Lomitapide is a synthetic lipid-lowering agent for oral administration. It is a microsomal triglyceride transfer protein inhibitor approved as Juxtapid® in US and as Lojuxta® in Europe as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis where available, to reduce low-density lipoprotein cholesterol (LDL-C), total cholesterol (TC), apolipoprotein B (apo B), and non-highdensity lipoprotein cholesterol (non-HDL-C) in patients with homozygous familial hypercholesterolemia (HoFH). The approved drug product is a mesylate salt of lomitapide, chemically known as N-(2,2,2-trifluoroethyl)-9-[4-[4-[[[4′(trifluoromethyl)[1,1′-biphenyl]-2-yl]carbonyl]amino]-1-piperidinyl]butyl]-9H-fluorene-9carboxamide methanesulfonate [“lomitapide mesylate” herein after] and has the structural formula

As per the approved label for Juxtapid® (US) “Lomitapide mesylate is a white to off-white powder that is slightly soluble in aqueous solutions of pH 2 to 5. Lomitapide mesylate is freely soluble in acetone, ethanol, and methanol; soluble in 2-butanol, methylene chloride, and acetonitrile; sparingly soluble in 1-octanol and 2-propanol; slightly soluble in ethyl acetate; and insoluble in heptane”.
As per Public Assessment Report for Lojuxta® (Europe) “Polymorphism has been observed for lomitapide mesylate. Of the different solid-state forms, hydrates, and solvates identified in the polymorph studies, only 2 desolvated solid-state forms, Form I and Form II, were identified in batches after drying to final drug substance.” The report further states, under the heading Manufacture, that “The final particle size distribution is controlled during the crystallisation step” (emphasis added) suggesting that the approved drug product lomitapide mesylate is a crystalline compound
U.S. Pat. No. 5,712,279 A discloses the lomitapide compound and a process for its preparation. It also discloses a process for preparation of lomitapide monohydrochloride.
U.S. Pat. No. 5,883,109 A discloses lomitapide mesylate specifically but no solid form was disclosed.
The reference article Synthesis and Applications of Isotopically Labelled Compounds, Vol. 8, Pg. 227-230 (2004) discloses the preparation of Deuterium labelled [d4]BMS-201038, [3H]BMS-201038, [14C]BMS-201038 wherein BMS-201038 is lomitapide mesylate.
International (PCT) Publication No. WO 2015/121877 A2 discloses lomitapide crystalline Form I and Form II as well as amorphous form of Lomitapide mesylate and processes for their preparation.
There is still a need to provide a novel polymorph of lomitapide or its salts which is suitable for pharmaceutical preparations. Therefore, the present invention provides new crystalline forms of lomitapide free base and lomitapide mesylate. The present invention also provides amorphous form of lomitapide free base and lomitapide mesylate, which is stable and useful for pharmaceutical preparations.
EXAMPLES
Example-1
Preparation of Lomitapide Mesylate
In a 250 mL round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 10 g lomitapide and 20 mL methanol were added and stirred to obtain a solution. 1.5 g methane sulfonic acid dissolved in 20 mL water was added slowly to the above solution under stirring. The reaction mixture was stirred till maximum salt formation was achieved. 50 mL water was added to the mixture, stirred for 15-20 min, filtered and washed with water. The product was dried further to obtain lomitapide mesylate.
EXAMPLE 2
Preparation of Amorphous Form of Lomitapide Mesylate
10 g lomitapide mesylate, 50 mL acetone and 150 mL ethyl acetate were heated in a 500 mL round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel at 50-55° C. and stirred to obtain clear solution. The solution was subjected to spray drying in JISL Mini spray drier LSD-48 with feed pump running at 30-35 rpm, inlet temperature 50-55° C., out let temperature 45-50° C., aspiration rate 1200-1300 rpm, hot air supply 1.8-2.2 Kg/cm2 and vacuum for conveying the dry product 80 mmHg. The product was collected from cyclone and characterized to an amorphous form by x-ray powder diffraction. The product was further dried to obtain the amorphous form of lomitapide mesylate
/////////////New patent, Lomitapide mesylate , Zydus Cadila Healthcare Ltd, US 20160083345, Amorphous