WO2017163257) PROCESS FOR PREPARING PURE LH-PYRAZOLO[3,4-D] PYRIMIDINE DERIVATIVE
IND-SWIFT LABORATORIES LIMITED
ARUL, Ramakrishnan; (IN).
SARIN, Gurdeep Singh; (IN).
WAS, Sandeep; (IN).
KUMAR, Vishal; (IN)
SARIN, Gurdeep Singh; (IN).
WAS, Sandeep; (IN).
KUMAR, Vishal; (IN)
The present invention relates to an efficient and industrially advantageous process for the preparation of pure lH-pyrazolo[3,4-d] pyrimidine derivative. In particular the present invention provides a process for the preparation of pure 4-amino-3-(4- phenoxyphenyl)-lH-pyrazolo[3,4-d] pyrimidine, a key intermediate of ibrutinib. Particularly, the present invention provides a process for the preparation of 3-amino-4-cyano-5-(4-phenoxy phenyl)pyrazole, wherein none of the intermediates have been isolated, an important precursor for the preparation of 4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-d] pyrimidine.
The present invention relates to an efficient and industrially advantageous process for the preparation of pure lH-pyrazolo[3,4-d] pyrimidine derivative. In particular the present invention provides a process for the preparation of pure 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d] pyrimidine, a key intermediate of ibrutinib, wherein none of the intermediates have been isolated to prepare 3-amino-4-cyano-5-(4-phenoxy phenyl)pyrazole, an important precursor.
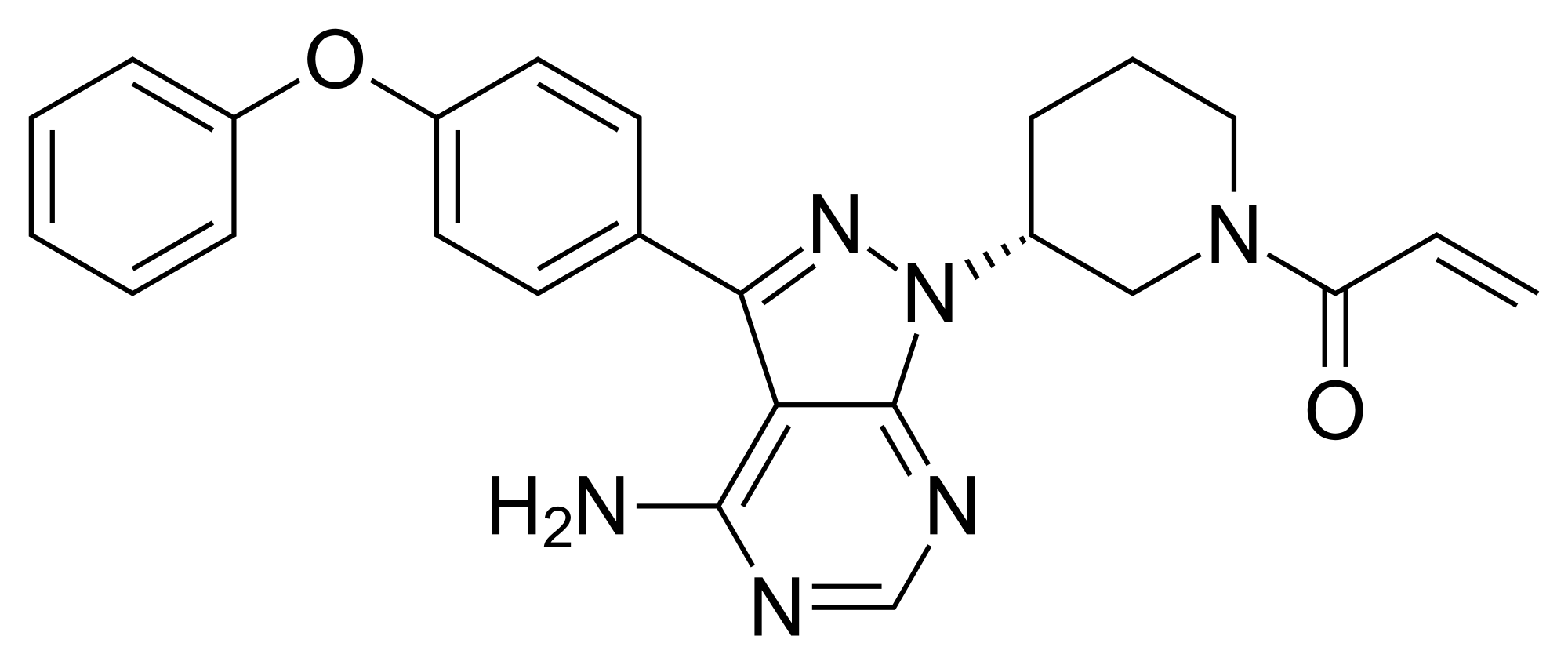
Ibrutinib (IMBRUVICA), chemically known as l-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin- 1 -yl]piperidin- 1 -yl] prop-2-en- 1 -one is an orally administered drug that targets Bruton’s tyrosine kinase (BTK). Ibrutinib may be used for treating both B cell-related hematological cancers/ B cell chronic lymphocytic leukemia, and autoimmune diseases such as rheumatoid arthritis, Sjogrens syndrome, lupus and asthma and is represented by following chemical formula:

Ibrutinib and its pharmaceutically acceptable salts were first disclosed in US patent US7,514,444. This patent discloses a process for the preparation of Ibrutinib by involving use of 4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-d]pyrimidine, as intermediate as shown below:


4-Amino-3-(4-phenoxyphenyl)- l H-pyrazolo[3,4-d]pyrimidine, a key intermediate of ibrutinib, and its preparation from 3-amino-4-cyano-5-(4-phenoxyphenyl) pyrazole was first disclosed in a PCT patent publication WO2001/019829 A2 as shown in below scheme.

Various other publications like US patents US7,514,444; US7.718,662; US8,883,803 and PCT publications WO2012/158843 A2; WO2013/010136A2 follow the same process for the preparation of 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidine as described above.
The process comprises the conversion of 4-phenoxybenzoic acid to the corresponding acid chloride, which is then taken up in mixture of toluene and tetrahydrofuran and further reacted with malononitrile in the presence of diisopropylethylethylamine in toluene. The reaction mixture is stirred overnight and after completion of reaction, followed by work up 1 , 1 -dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene is isolated as a residue and which is further purified.
The resulting l, l-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene is reacted with trimethylsilyldiazomethane in a mixture of acetonitrile and methanol in the presence of diisopropylethylamine as a base. The resulting reaction mixture is stirred for 2 days to give l, l-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (O-methylated product) as an oil, which is purified by flash chromatography.
The O-methylated product is treated with hydrazine hydrate to give 3-amino-4-cyano- 5-(4-phenoxyphenyl)pyrazole, which is further reacted with formamide at a temperature of 180°C to give 4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4- d]pyrimidine as pale brown-grey solid.
Since, the above process involves the isolation of intermediates and takes long time during reaction completion. Therefore, it is lengthy, not efficient. Further publication is silent about the purity of 4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-d]pyrimidine. Acetonitrile solvent has been used in methylation reaction, which is carcinogenic.
The cyclization reaction has been carried out at 180°C and it is observed that the cyclization reaction at high temperature of 180°C, results in grey brown solid colour of 4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-d]pyrimidine, may be due to presence of inorganic impurities.
The said process also requires the use of expensive (trimethylsilyl)diazomethane to obtain O-methylated product, which is sensitive to air and water, and hence, the methylation reaction has to be carried out in the absence of water, under anaerobic conditions; silica and flash chromatography are also used for purifying O-methylated product. Since the above process involves complicated operation processes, which leads to high production cost and therefore is not an attractive option at industrially scale.
PCT publication WO2014/173289A1 discloses a process for preparation of 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as shown below and its conversion into 4- amino-3-(4-phenoxy phenyl)- lH-pyrazolo[3,4-d]pyrimidine has not been disclosed.

The process involves conversion of 4-phenoxybenzoic acid to the corresponding acid chloride, followed by reaction with malononitrile in the presence of diisopropylethylethylamine in tetrahydrofuran. The reaction mixture has been stirred for 16 hours and thereafter l, l -dicyano-2-hydroxy-2-(4-phenoxyphenyl) ethene is isolated from reaction mixture. A solution of l, l-dicyano-2-hydroxy-2-(4- phenoxyphenyl)ethene in trimethoxymethane has been heated for 16 hours to give l, l-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (O-methylated product), which is then reacted with hydrazine hydrate to give 3-amino-4-cyano-5-(4-phenoxy phenyl)pyrazole.
The above process is inefficient, since it involves isolation of intermediates and takes long time to complete the reactions and purity of 3-amino-4-cyano-5-(4- phenoxypheny pyrazole has not been disclosed.
A similar approach has been described in a PCT publication WO2014/082598 A 1 for preparation of 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole and is presented as below:

The process involves conversion of 4-phenoxybenzoic acid to the corresponding acyl chloride by using sulfurous dichloride, followed by reaction with malononitrile in the presence of sodium hydride to obtain l, l-dicyano-2-hydroxy-2-(4-phenoxy phenyl)ethene, which is recrystallized from 1,4-dioxane. The hydroxy moiety is then methylated using dimethyl sulphate to give l, l-dicyano-2-methoxy-2-(4-phenoxy phenyl)ethene (O-methylated product) which is recrystallized from a mixture of hexane and ethylactetate. The solution of resulting O-methylated product in ethanol was treated with hydrazine hydrate at reflux temperature to give 3-amino-4-cyano-5- (4-phenoxy phenyl)pyrazole, followed by its recrystallization in hexane and further, its conversion into 4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-d]pyrimidine was not disclosed.
The above process also involves isolation of intermediates; their purification which leads to longer time in reaction completion, and it does not disclose the purity of 3- amino-4-cyano-5-(4-phenoxyphenyl)pyrazole. Further the above process involves use of sodium hydride, which is a hazardous reagent and can ignite in air during scale up. Several alternative methods have been reported in literature, wherein process for the preparation of 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidine has been disclosed and are discussed herein.
A Chinese patent application CN103121999A discloses a process of preparation of 4- amino-3 -(4-phenoxy phenyl)- 1 H-pyrazolo[3 ,4-d] pyrimidine, as below :

The process involves reaction of 3-bromo-lH-pyrazolo[3,4-d]pyrimidin-4-amine with (4-phenoxyphenyl)boronic acid in the presence of alkali agents and aprotic solvents to give 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidine.
The said Chinese application is also silent about the purity of target compound and even starts with the advance intermediates, which are expensive and make the process unattractive from industrial point of view.
A similar approach has been described in US patent US8,940,893; PCT publication WO2013/1 13097A1 and WO2015/018333 A 1 for preparing 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidine .
In US patent US8,940,893 and PCT publication WO2013/1 13097A1, 4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-d]pyrimidine is purified by using Combi-flash chromatography on silica gel. In PCT publication WO2015/018333A1, 4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-d]pyrimidine is purified by recrystallization in ethyl acetate.
The purity of 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidine has not been reported in above publications too. Further two of the above processes involve tedious step of chromatographic purification, which is not industrial viable.
Another Chinese patent application CN 103965201 A discloses a process for the preparation of 4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-d]pyrimidine, wherein 3-bromo-lH-pyrazolo[3,4-d]pyrimidin-4-amine was reacted with trimethyl tin (4- phenoxy phenyl) to give 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4- d]pyrimidine and followed by its recrystallization in isopropanol, as shown below:

The said Chinese application is also silent about the purity of 4-amino-3-(4- phenoxyphenyl)- lH-pyrazolo[3,4-d]pyrimidine and is not cost-effective because it starts with advance intermediates, which are expensive. Therefore, said route of synthesis is not industrially applicable.
Purity of an API as well as intermediates is of great importance in the field of pharmaceutical chemistry. It is well documented in the art that direct product of a chemical reaction is rarely a single compound with sufficient purity to comply with pharmaceutical standards. The impurities that can be present in pharmaceutical compounds are starting materials, by-products of the reaction, products of side reactions, or degradation products.
According to ICH guidelines, process impurities should be maintained below set limits by specifying the quality of raw materials, their stoichiometric ratios, controlling process parameters, such as temperature, pressure, time and including purification steps, such as crystallization, distillation and liquid-liquid extraction etc., in the manufacturing process. Typically, these limits should less than about 0.15 % by weight of each identified impurity. Limits for unidentified and/or uncharacterized impurities are obviously lower, typically less than 0.10 % by weight. The limits for genotoxic impurities could be much lower depending upon the daily dose of the drug and duration of the treatment. Therefore, in the manufacture of a drug substance, the purity of the starting materials is also important, as impurities may carry forward to the active pharmaceutical ingredient such as ibrutinib.
In view of the above, most of the prior art processes involve isolation of intermediates, additional purification steps and silent about the purity or the assay of 4-amino-3-(4-phenoxy phenyl)- lH-pyrazolo[3,4-d]pyrimidine.
Thus, there is an urgent need for the development of a synthetic process which produces pure 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidine or its acid addition salts.
The present invention fulfills the need in the art and provides an improved, industrially advantageous process for the synthesis of pure 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d] pyrimidine, a key intermediate in the preparation of ibrutinib, through preparation of 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole from 4-phenoxy benzoic acid using same organic solvent and none of the intermediates have been isolated.
Examples:
Example 1: Preparation of 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole
4-Phenoxybenzoic acid (200g) was slowly added to thionyl chloride (400ml) at a temperature of 25-30°C and resulting reaction mixture was heated under stirring to a temperature of 60-65°C for 5 hours. Thionyl chloride was distilled off under vacuum at temperature below 60°C. Toluene (2x400ml) was added to the resulting oily residue and thereafter distilled out completely under vacuum below 60°C to remove traces of thionyl chloride to obtain 4-phenoxybenzoyl chloride as a viscous oil. The resulting viscous oil of 4-phenoxybenzoyl chloride was dissolved in toluene (2000ml). Malononitile (80g) and diisopropylethylamine (320ml) were sucessively added to the reuslting solution at a temperature of 25-30°C slowly, maintaining reaction temperature 50-55°C. The reaction mass was further stirred for 30 minutes. After completion of the reaction, the reaction mass was cooled to 25-30°C and a solution of sulfuric acid ( 1.25 M) was added. The reaction mixture was then stirred at a temperature of 25-30°C for 30 minutes, and the layers were separated. The organic layer was washed with a solution of sodium chloride ( 10%) and the resulting organic layer was used directly in next reaction.
Dimethyl sulfate (200ml) and sodium bicarbonate (200g) were added to the resulting organic layer at a temperature of 25-30°C. Thereafter, temperature of reaction mass was raised to 80-90°C and reaction mass was stirred for 1-2 hours. After completion of reaction, the reaction mass was cooled to a temperature of 25-30°C, demineralized water (2000ml) was added and stirred for 10-15 minutes. The layers were separated and the aqueous layer was extracted with toluene (1000ml). All the organic layers were combined and washed with sodium chloride solution ( 10%). Activated carbon (20g) was added and reaction mixture was stirred for 30 minutes. The solution was filtered through hyflo bed and to the resulting organic layer hydrazine hydrate ( 120ml) was added at a temperature of 25-30°C. During the addition exothermicity was observed, and temperature of the reaction mass was rose up to 40-50°C. Thereafter, the reaction mass was stirred at a temperature of 25-30°C for 1 -2 hours. The resulting precipitated solid was filtered, slurry washed with dichloromethane (400ml) and finally, dried to obtain title compound of formula V ( 140g) purity 93.28% measured by HPLC.
Example 2: Preparation of 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole
4-Phenoxybenzoic acid (lOOg) was slowly added to thionyl chloride (200ml) at a temperature of 25-30°C and resulting reaction mixture was heated under stirring to a temperature of 50-55°C for 5 hours. Thionyl chloride was distilled off under vacuum at temperature below 50°C. Toluene (250ml) was added to the resulting oily residue and thereafter distilled out completely under vacuum below 50°C to remove traces of thionyl chloride to obtain 4-phenoxybenzoyl chloride as a viscous oil. The resulting viscous oil of 4-phenoxybenzoyl chloride was dissolved in toluene (500ml). Malononitile (35.58ml) and diisopropylethylamine (160ml) were sucessively added to the reuslting solution at a temperature of 25-30°C slowly, maintaining reaction temperature around 50-55°C. The reaction mass was further stirred for 10 minutes. After completion of the reaction, the reaction mass was cooled to 25-30°C and a solution of sulfuric acid (70 ml in 1000 ml water) was added. The reaction mixture was then stirred at a temperature of 25-30°C for 30 minutes, and the layers were separated. The organic layer was washed with a solution of sodium chloride (10%) and the resulting organic layer was used directly in next reaction.
Dimethyl sulfate (95.1 1ml) and sodium bicarbonate (96.16g) were added to the resulting organic layer at a temperature of 25-30°C. Thereafter, temperature of reaction mass was raised to 80-90°C and reaction mass was stirred for 1-2 hours. After completion of reaction, the reaction mass was cooled to a temperature of 55- 60°C, demineralized water ( 1000ml) was added. The reaction mass was cooled to a temperature of 25-30°C and stirred for 10- 15 minutes. The layers were separated and the aqueous layer was extracted with toluene (500ml). All the organic layers were combined and washed with sodium chloride solution ( 10%). To the resulting organic layer hydrazine hydrate (50ml) was added at a temperature of 25-30°C. During the addition exothermicity was observed, and temperature of the reaction mass was rose up to 40-45°C. Thereafter, the reaction mass was stirred at a temperature of 25-30°C for 1-2 hours. The resulting precipitated solid was filtered, suck dried to obtain 3- amino-4-cyano-5-(4-phenoxyphenyl)pyrazole compound of formula V ( 123g) purity 86.96% measured by HPLC.
Example 3: Purification of 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (36g) was suspended in isopropanol (350ml) and temperature of the reaction mixture was raised and allowed to reflux to dissolve the solid completely to provide a clear solution. Then, solvent was distilled off under vacuum to obtain a residue and isopropanol (50ml) was added and after stirring for hours the solid was filtered and dried to afford 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole compound of formula V (26g) and having purity of 97.54 % by HPLC .
Example 4: Purification of 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (36g) was suspended in isopropanol (350ml) and temperature of the reaction mixture was raised upto reflux to dissolve the solid completely upto clear solution. Water (1050ml) was added to the solution and the reaction mixture was gradually cooled to crystallize the product. The resulting solid was filtered, washed with two volumes of isopropanol, dried in vacuum oven at a temperature of 40-45 °C to afford 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole compound of formula V (20g) and having a HPLC purity of 97.23% .
Example 5: Preparation of pure 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4- d] pyrimidine compound of formula I
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (20g) was suspended in formamide (100 ml) and heated at a temperature of 130°C, after completion of reaction, the reaction mixture was cooled to a temperature of 30-35°C and demineralized water (500ml) was added and the reaction mixture was stirred at a temperature of 25-30°C for 45 minutes. The resulting solid was filtered and acetone (200ml) was added stirred the reaction mixture for 30-45 minutes. The resulting solid was filtered, washed, dried to afford pure 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidine compound of formula 1 (12g) having purity 99.6% measured by HPLC.
Example 6: Preparation of pure 4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4- d] pyrimidine compound of formula I
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (lOOg) was suspended in formamide (500ml) and heated at a temperature of 135-140°C, after completion of reaction, the reaction mixture was cooled to a temperature of 30-35°C and demineralized water (1000ml) was added and the reaction mixture was stirred at a temperature of 20-25°C for 1 hour. The resulting solid was filtered, washed with water (500ml) then successively slurry washed with toluene (2 x 500ml) and dried to afford pure 4- amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4-d] pyrimidine compound of formula I
(70g) having purity 99.8% measured by HPLC; assay > 98%; residue on ignition 0.05%; heavy metals < 20ppm.
Example 7: Preparation of (lS)-l-[(3R)-3-piperidyl]-3-(p-phenoxyphenyl)-l,2,5,7-tetraza-lH-inden-4-ylamine
Diisopropyl diazodicarboxylate (DAID, 1.2 ml,) was added to a solution of 1-tert-butyloxycarbonyl-3-(S)-hydroxypiperidine ( l .Og,) and triphenylphosphine (2.59g) in tetrahydrofuran (50.0ml). To the resulting yellow solution, 3-(p-phenoxyphenyl)-l ,2,5,7-tetraza- lH-inden-4-ylamine (l .Og). was added and warmed till dissolution, and stirred overnight at room temperature. The reaction mixture was filtered and the solvent was distilled under vacuum to get an oily residue, which was further purified by flash chromatography (30-50 % ethyl acetate/ hexane) on silicagel to give 0.3 g (0.3 w/w) of tert-butyloxycarbonyl-( l S)- l-[(3R)-3-piperidyl]-3-(p-phenoxyphenyl)- l,2,5,7-tetraza- lH-inden-4-ylamine as a light brown solid. The resulting solid was dissolved in dichloromethane (5 ml) and trifluoroacetic acid (0.6 ml) was added to it. After completion of reaction, water was added to reaction mass, followed by addition of methyl tert-butyl ether (20.0 ml). The layers were separated and the aqueous layer was basified with potassium carbonate and extracted with dichloromethane (15.0 ml x 2). The organic layer dried over sodium sulfate, filtered and evaporated to yield 0.2 g (0.6 w/w) of title compound as light yellow oil.
Example 8: Preparation of l-(3-(4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo [3,4- d]pyrimidin-l-yl)piperidin-l-yI)prop-2-en-l-one (Ibrutinib)
To a solution of acryloyl chloride (0.06g) in tetrahydrofuran (15.0 ml), a mixture of triethylamine (O. lg) and (lS)-l-[(3R)-3-piperidyl]-3-(p-phenoxyphenyl)-l,2,5,7- tetraza- lH-inden-4-ylamine (0.2g) in tetrahydrofuran (7.8 ml) was added. The reaction mixture was stirred at 25-30°C for 18 hous and filtered. The solvent was removed under vacuum to obtain crude ibrutinib, which was further purified by column chromatography on silica gel to obtain pure ibrutinib as crystalline solid.
Formula VI

Formula VII

Formula I

Formula II

Formula III
Formula IV
Formula V

///////WO 2017163257, NEW PATENT, IBRUTINIB, IND-SWIFT LABORATORIES LIMITED