US 8350029
| Inventors | Dharmaraj Ramachandra Rao, Rajendra Narayanrao Kankan, Srinivas Laxminarayan Pathi, |
| Original Assignee | Cipla Limited |
CIPLA Limited, Mumbai, India

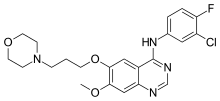
Gefitinib is an anilinoquinazoline which is useful in the treatment of a certain type of lung cancer (non-small cell lung cancer or NSCLC) that has not responded to chemotherapy. The chemical name for gefitinib is 4-(3′-chloro-4′-fluoroanilino)-7-methoxy-6-(3-morpholinopropoxy)quinazoline. Its structural formula is:
 The earliest known synthesis of gefitinib was first disclosed in the patent application WO 96/33980. The synthetic method employed is depicted in the following reaction scheme 1.
The earliest known synthesis of gefitinib was first disclosed in the patent application WO 96/33980. The synthetic method employed is depicted in the following reaction scheme 1.
 The process involves selective demethylation of 6,7-dimethoxy quinazoline-4-one using methanesutfonic acid and L-methionine to get its 6-hydroxyl derivative, which is protected by acetylation. The acetoxy compound is chlorinated and condensed with chloro-fluoroaniline. Hydrolysis of the acetoxy compound followed by etherification with 3-morpholinopropyl chloride gives crude gefitinib which is purified by column chromatography. The process suffers from many disadvantages as it involves several protection and deprotection steps. The selective demethylation using methionine results in isomeric impurities and has to be purified or else the impurity carries over to subsequent steps in the preparation of gefitinib making it more difficult to isolate a pure product. The process also leads to formation of an N-alkylated impurity at the final stage which must be separated by column chromatography to obtain gefitinib.
The process involves selective demethylation of 6,7-dimethoxy quinazoline-4-one using methanesutfonic acid and L-methionine to get its 6-hydroxyl derivative, which is protected by acetylation. The acetoxy compound is chlorinated and condensed with chloro-fluoroaniline. Hydrolysis of the acetoxy compound followed by etherification with 3-morpholinopropyl chloride gives crude gefitinib which is purified by column chromatography. The process suffers from many disadvantages as it involves several protection and deprotection steps. The selective demethylation using methionine results in isomeric impurities and has to be purified or else the impurity carries over to subsequent steps in the preparation of gefitinib making it more difficult to isolate a pure product. The process also leads to formation of an N-alkylated impurity at the final stage which must be separated by column chromatography to obtain gefitinib.
Several other approaches are also described in the literature to make gefitinib.
WO 2004/024703 discloses a process for the preparation of gefitinib starting from 3-hydroxy-4-methoxy benzonitrile which involves condensation of 3-hydroxy-4-methoxy benzonitrile with morpholino propyl chloride, nitration, reduction with sodium dithionite to amino compound, hydrolysis of nitrile to amide, cyclisation in the presence of formamide to obtain quinazoline, chlorination with phosphorous oxychloride and finally condensation with chloro-fluoro aniline to obtain gefitinib. The process involves multiple steps and hence is time consuming.
WO 2005/023783 discloses a process for the manufacture of gefitinib starting from 2-amino-4-methoxy-5-(3-morpholinopropoxy)benzonitrile. The process involves a rearrangement reaction of 3-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)3,4-dihydroqunazoline-4-imine. The process is not feasible industrially, as the basic raw material is not readily available on a commercial scale and involves the use of excess 3-chloro-4-fluoroaniline which is expensive. A further draw back of the process is in the isomerization of the 4-imine compound which requires anhydrous conditions at high temperature for a longer duration of 96 hours. All the problems associated with this prior art process are overcome by the novel process of the present invention.
WO2005/070909 discloses a process for the preparation of gefitinib starting from isovanillin as depicted in scheme 2
 The WO' 909 process has disadvantages as it forms cis-trans geometrical isomers of the oxime, which have different reactivities. Furthermore, the process uses a large excess of acetic anhydride to convert the oxime to the nitrile at higher temperature.
The WO' 909 process has disadvantages as it forms cis-trans geometrical isomers of the oxime, which have different reactivities. Furthermore, the process uses a large excess of acetic anhydride to convert the oxime to the nitrile at higher temperature.
The patent applications 901/CHE/2006 and 903/CHE/2006 disclose another route for preparing gefitinib starting from isovanillin. The process involves formation of a formamido compound [N′-[2-cyano-4-{3-(4-morpholinyl)propoxy}phenyl]-N,N-dimethyl formamide], which is unstable and may result in undesired impurities in the final condensation with 3-chloro-4-fluoro aniline, thereby making the process less feasible on an industrial scale.
The processes disclosed in the prior art are cumbersome. Therefore, there exists a need for a more economical and efficient method of making gefitinib which is suitable for industrial scale-up.
The process of the present invention avoids use of reagents such as sodium dithionite, acetic anhydride and allows substantial reduction in the number of problems associated with these reagents.


Several other approaches are also described in the literature to make gefitinib.
WO 2004/024703 discloses a process for the preparation of gefitinib starting from 3-hydroxy-4-methoxy benzonitrile which involves condensation of 3-hydroxy-4-methoxy benzonitrile with morpholino propyl chloride, nitration, reduction with sodium dithionite to amino compound, hydrolysis of nitrile to amide, cyclisation in the presence of formamide to obtain quinazoline, chlorination with phosphorous oxychloride and finally condensation with chloro-fluoro aniline to obtain gefitinib. The process involves multiple steps and hence is time consuming.
WO 2005/023783 discloses a process for the manufacture of gefitinib starting from 2-amino-4-methoxy-5-(3-morpholinopropoxy)benzonitrile. The process involves a rearrangement reaction of 3-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)3,4-dihydroqunazoline-4-imine. The process is not feasible industrially, as the basic raw material is not readily available on a commercial scale and involves the use of excess 3-chloro-4-fluoroaniline which is expensive. A further draw back of the process is in the isomerization of the 4-imine compound which requires anhydrous conditions at high temperature for a longer duration of 96 hours. All the problems associated with this prior art process are overcome by the novel process of the present invention.
WO2005/070909 discloses a process for the preparation of gefitinib starting from isovanillin as depicted in scheme 2

The patent applications 901/CHE/2006 and 903/CHE/2006 disclose another route for preparing gefitinib starting from isovanillin. The process involves formation of a formamido compound [N′-[2-cyano-4-{3-(4-morpholinyl)propoxy}phenyl]-N,N-dimethyl formamide], which is unstable and may result in undesired impurities in the final condensation with 3-chloro-4-fluoro aniline, thereby making the process less feasible on an industrial scale.
The processes disclosed in the prior art are cumbersome. Therefore, there exists a need for a more economical and efficient method of making gefitinib which is suitable for industrial scale-up.
The process of the present invention avoids use of reagents such as sodium dithionite, acetic anhydride and allows substantial reduction in the number of problems associated with these reagents.
Process for Preparation of Gefitinib
Gefitinib, 66, is used in the treatment of certain types of lung cancer, and a number of methods are reported for its synthesis. These are described as cumbersome and can require excessive amounts of reagents or involve difficult purification methods. Some processes use reagents such as sodium dithionite or Ac2O, and these are said to create problems. This patent discloses two routes for the synthesis of 66 that are claimed to avoid such problems. The first route, shown in Scheme 22, is the subject of the claims of the patent and starts with the nitration of isovanillin 57ain HOAc to give 57b that is recovered in 65% yield. Treatment of 57b with 58 produces 59a that is isolated in 92% yield, and this is then oxidised with H2O2 to form the acid 59b that is isolated in 86% yield. Reduction of the nitro group is then carried out to give 60, and there are three methods described for this reaction. The first is catalytic hydrogenation with Pd/C that gives a 90% yield of60. The reaction pressure is reported as being 5–6 kg, a common term in India used as short-hand for the pressure unit of kg/m2. Reduction using H2NNH2 in the presence of FeCl3, Al2O3, and charcoal gives a 83.6% yield of 60. In a hydrogen-transfer reaction with HCO2NH4 and Pd/C compound, 60 is recovered in 84.5% yield. The cyclisation of 60 to form 61 is carried out in a Niementowski reaction using HCO2NH4 and HCO2NH2, and the product is recovered in 90% yield. Reaction of 61 with SOCl2 produces 62, and this is isolated in 95% yield. Only the main reagents are shown in the scheme, and workup details are omitted.

Scheme 22. a
aReagents and conditions: (a) (i) HNO3, HOAc, −5 °C; (ii) 30 °C, 12 h. (b) K2CO3, MeCN, reflux, 4 h. (c) (i) 30% NaOH/MeOH, 45 °C; (ii) add 35% H2O2 over 4 h, 45 °C, pH 11. (d) Pd/C, H2, EtOAc, 40 °C, 4 h. (e) HCO2NH4, HCO2NH2, 180 °C, 4 h. (f) SOCl2, DMF, reflux, 8 h.
In the next stage of the synthesis, shown in Scheme 23, compound 62 is reacted with morpholine63 to give 64 in 85% isolated yield. In the final step 64 is reacted with 65 to produce 66 that is recovered in 70% yield (purity not reported).

Scheme 23. a
aReagents and conditions: (a) (i) 75 °C, 8 h; (ii) cool to rt, add H2O; (iii) separate extract in DCM, H2O wash, dry, evaporate. (b) MeOH, 30 °C, 0.25 h; (ii) add 65, reflux 6 h; (iii) add HCl at 20 °C; (iii) <10 °C, 0.5 h; (iv) filter, MeOH wash; (v) dissolve in PhMe/MeOH, concentrate; (vi) cool <10 °C, filter, PhMe wash, dry.
The patent also describes an alternative route to 66 that is outlined in Schemes 24 and 25although it is not covered by the patent claims. The route starts with the oxidation of 57a using H2O2 to give the acid 67a that is esterified to form 67b that is isolated in 83% yield. Nitration of67b with HNO3 in HOAc produces 68a that is isolated in 74% yield and then reduced to 68b over Pd/C. The amine 68b is recovered in 93% yield and then reacted with 69 to give the quinazoline70a that is recovered in 92% yield and then acetylated to form 70b. There is no example describing this acetylation nor are there any for the remaining steps of this route shown in Scheme25, and the reactions are just generally referred to in the text.

Scheme 24. a
aReagents and conditions: (a) (i) 30% NaOH/MeOH, 45 °C; (ii) add 35% H2O2 over 3 h, 45 °C, pH 11. (b) 10% HCl/MeOH, reflux, 6 h. (c) 70% HNO3, HOAc, −5 °C, 18 h. (d) Pd/C, H2, EtOAc, 40 °C, 4 h. (e) MeOH, reflux, 10 h. (f) No details.

Scheme 25. a
aReactions: (a) Chlorination. (b) Condensation. (c) Hydrolysis. (d) Coupling.
The examples report experiments carried out on a reasonable scale with some producing up to 200 g of products. Unfortunately, there are no details of the purity of any of the intermediates, and although the patent states that the desired final product 66 is purified by acid/base treatment or crystallisation, there are no details provided.
Advantages
The process does avoid the use of some difficult reagents used elsewhere, but whether the process gives a higher-purity product than alternatives is not clear.
scheme 3.


scheme 4.


EXAMPLE 1 Preparation of 4-(3′-chloro-4′-fluoroanilino)-7-methoxy-6-(3-morpholinopropoxy)-quinazoline (gefitinib) (Formula I)Methanol (1200 ml) and 6-(3-morpholino propoxy)-7-methoxy-4-chloro quinazoline (200 gm) were stirred for 15 minutes at 25-30° C., then a solution of 4-fluoro-3-chloroaniline in methanol (213 gm in 400 ml) was charged and refluxed for 6 hours. The reaction mass was cooled to 15-20° C., hydrochloric acid (40 ml) was added drop wise, and stirred at 5-10° C. for 30 minutes. The solid obtained was filtered and washed with chilled methanol (150 ml). The solid was dissolved in a mixture of toluene (30 volume) and methanol (5 volume), the reaction mass was concentrated to half the volume and cooled to 5-10° C. The solid obtained was filtered, washed with toluene (200 ml) and dried at 45-50° C. to yield the title compound (183 gm, 70% yield).
EXAMPLE 2 Preparation of 6-(3-morpholino propoxy)-7-methoxy-4-chloroquinazoline (Formula VII)DMF (3 lt), 6-(3-chloropropoxy)-7-methoxy-4-chloro quinazoline (200 gm) and morpholine (210 gm), were heated to 70-75° C. for 6-8 hours. The reaction mass was cooled to room temperature, and methylene chloride (2.5 lt) and water (2.5 lt) were charged. The layers separated and the aqueous layer extracted with methylene chloride twice (500 ml). The combined methylene chloride layer was washed with water, dried over sodium sulphate (10 gm) and concentrated completely at 35-40° C. to yield the title compound (200 gm, 85% yield).
EXAMPLE 3 Preparation of 6-(3-chloropropoxy)-7-methoxy-4-chloroquinazoline (Formula VI)6-(3-chloropropoxy)-7-methoxyquinazoline-4-one (400 gm), thionyl chloride (3.2 lt) and DMF (100 ml) were refluxed for 7-8 hours. Thionyl chloride was distilled off completely under reduced pressure below 45° C. Methylene chloride (2.5 lt) and water (1.5 lt) were charged, stirred for 30 minutes at room temperature and the layers separated. The aqueous layer was extracted twice with methylene chloride (500 ml), the combined methylene chloride layer was washed with 1% sodium bicarbonate solution (1 lt), dried over sodium sulphate (20 gm) and concentrated under reduced pressure at 35-40° C. The residue was stirred with isopropyl alcohol (400 ml) at 40-45° C. for 1 hour, cooled to 0-5° C., the solids filtered, washed with chilled isopropyl alcohol (200 ml) and dried under vacuum at 45° C. to yield the title compound (406 gm, 95% yield).
EXAMPLE 4 Preparation of 6-(3-chloropropoxy)-7-methoxyquinazoline-4-one (Formula V)2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid (450 gm), formamide (2250 ml) and ammonium formate (200 gm) were heated to 170-180° C. for 3-4 hours. The reaction mass was concentrated under reduced pressure at 140-150° C. The residue was stirred in methanol (1000 ml) at 45-50° C. and cooled to 5-10° C. The solid obtained was filtered to yield the title compound (420 gm, 90% yield).
EXAMPLE 5 Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid (Formula IV) a) Preparation of 3-(3-chloropropoxy)-4-methoxy-6-nitrobenzoic acidMethanol (4 lt), 3-(3-chloropropoxy)-4-methoxy-6-nitro benzaldehyde (560 gm) and 30% methanolic NaOH solution (5 ml) were heated to 45° C. To this reaction mass 35% of H2O2 solution (1200 ml) was added drop wise in 3-4 hours maintaining a pH of 10.5-11.5 with 30% methanolic NaOH solution. The reaction mass was quenched into ice water (10 kg) and the pH adjusted to 2.0-3.0 using hydrochloric acid. The solid obtained was filtered, washed with 50% aqueous methanol (500 ml) and dried at 45-50° C. to yield the title compound (510 gm, 86% yield).
bi) Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid—Using Hydrogen GasEthyl acetate (3 lt), Pd/C (50 gm) and 3-(3-chloropropoxy)-4-methoxy-6-nitrobenzoic acid (500 gm) were hydrogenated under a hydrogen pressure of 5-6 kg at 35-40° C. for 3-4 hours. The reaction mass was filtered and the clear filtrate was distilled under reduced pressure at 45-50° C. To the residue, hexane (1 lt) was charged, stirred at room temperature, the solids filtered and dried at 45-50° C. to yield the title compound (403 gm, 90% yield).
(bii) Preparation of 2-amino-4methoxy-5-(3-chloropropoxy)benzoic acid—Using Hydrazine Hydrate3-(3-chloropropoxy)-4-methoxy-6-nitrobenzoic acid (100 gm), hydrazine hydrate (50 gms), neutral alumina (20 gms), charcoal (10 gms), water (50 ml) and methanol (500 ml) were mixed together. The reaction mass was heated to 50° C. A solution of ferric chloride (2 gms, 0.012M) in 50 ml methanol was introduced slowly at 55-60° C. The reaction mass was filtered over hyflo and the clear filtrate evaporated. The residue obtained was dissolved in 1.0-lit ethyl acetate, washed organic extract with water, evaporated to obtain title compound. (75 gms, 83.6%)
(biii) Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid—Using Ammonium Formate3-(3-chloropropoxy)-4-methoxy-6-nitro benzoic acid (165 gms), 5% Paladium on carbon (16.5 gms) and DMF (0.66 lit) were mixed together. The reaction mass was heated to 40° C. Ammonium formate (82.5 gms) was charged in lots maintaining temperature below 50° C. The temperature of reaction mass slowly raised to 70° C. and maintained for 2 hours. The reaction mass was cooled to 30° C. and catalyst was removed by filtration and the clear filtrate evaporated. The residue was dissolved in ethyl acetate (0.825 lit), washed with water and evaporated to yield the title compound. (125 gms, 84.5%)
EXAMPLE 6 Preparation of 3-(3-chloropropoxy)-4-methoxy-6-nitro benzaldehyde (Formula III)5-nitro isovanillin (500 gm), acetonitrile (3.5 lts), K2CO3 (750 gm) and chlorobromopropane (780 gm) were refluxed for 4 hours. The reaction mass was filtered hot, washed with acetonitrile (1 lt) and the filtrate was distilled off to remove solvent. The residue was dissolved in methylene chloride (4 lt) and washed with water. Water (3 lt) was charged to the methylene chloride layer, the pH adjusted to 7.0 to 7.5 with acetic acid, the methylene chloride layer separated, dried over sodium sulphate (50 gm) and distilled out completely under reduced pressure below 40° C. The residue was stirred with 2 volumes of n-Hexane at 40-45° C., cooled slowly to 0-5° C., the solids filtered, washed with n-Hexane (250 ml) and dried at 40-45° C. to yield the title compound (638 gm, 92% yield).
EXAMPLE 7 Preparation of 5-nitro isovanillin (Formula II)Isovanillin (500 gm) and acetic acid (1750 ml) were cooled to −5 to 0° C. To this solution, nitric acid (750 ml) was charged slowly at −5 to 0° C. with stirring. The temperature of the reaction mass was slowly raised to 25-30° C. and maintained for 12 hours. The reaction mass was quenched into ice water (4 kg), the solids filtered and washed with water (2 lt). The solids were stirred with a 1% sodium bicarbonate solution (1 lt), filtered and dried at 45-50° C. The solid was dissolved in 6 volumes of ethyl acetate, ethyl acetate was distilled off up to half the volume and 3 volumes of n-Hexane were charged slowly at 45-50° C. The reaction mass was cooled slowly to 0-5° C., maintained for 1 hour, the solids filtered, washed with 0.5 volumes of 1:1 mixture of ethyl acetate:n-Hexane and dried at 45-50° C. to yield the title compound (423 gm, 65% yield).
EXAMPLE 8 Preparation of Methyl-2-hydroxy-3-methoxy benzoate (Formula VIII) a) Preparation of 3-hydroxy-4-methoxy benzoic acidMethanol (350 ml), isovanillin (50 gm) and 30% methanolic sodium hydroxide solution (1 ml), were heated to 45° C. To this solution, 35% hydrogen peroxide solution (107 ml) was charged slowly maintaining pH at 10.5 to 11.5 using methanolic sodium hydroxide solution over a period of 2-3 hours. The reaction mass was quenched into chilled water (1 lt) and the pH adjusted to 2-3 using hydrochloric acid. The solids were filtered, washed with 50% aqueous methanol (50 ml) and dried at 45-50° C. to yield 3-hydroxy-4-methoxy benzoic acid.
b) Preparation of Methyl-2-hydroxy-3-methoxy benzoateThe solid obtained in step a), was refluxed with 10% methanolic hydrochloric acid solution (250 ml) for 6 hours. The reaction mass was quenched into chilled water (1 lt) and repeatedly extracted with methylene chloride (250 ml). The combined methylene chloride layer was washed with water (100 ml×2) and methylene chloride distilled out completely at 35-40° C. The residue was stirred in hexane (1.50 ml), at 25-30° C. The solid obtained was filtered, washed with: hexane (25 ml) and dried at 40-45° C. to yield the title compound (50 gm, 83% yield).
EXAMPLE 9 Preparation of Methyl-5-hydroxy-4-methoxy-2-nitro benzoate (Formula IX)Methyl-2-hydroxy-3-methoxy benzoate (50 gm) and acetic acid (175 ml) were cooled to 0-5° C. To this solution, 70% nitric acid solution (75 ml) was charged slowly at 0-5° C. under stirring and the reaction mass was further stirred for 18 hours. The reaction mass was quenched into chilled water (800 ml) and extracted repeatedly with methylene chloride (400 ml). The combined methylene chloride layer was washed with water, followed by 1% potassium carbonate solution (100 ml), dried over sodium sulphate and methylene chloride distilled off completely at 35-40° C. The residue was dissolved in 10% aqueous methanol (250 ml). The filtrate was gradually cooled to 0-5° C. and maintained for 1 hour. The solid obtained was filtered, washed with 10% aqueous methanol (100 ml) and dried at 40-45° C. to yield the title compound (46 gm, 74% yield).
EXAMPLE 10 Preparation of Methyl-2-amino-5-hydroxy-4-methoxy benzoate (X)Ethyl acetate (300 ml), methyl-5-hydroxy-4-methoxy-2-nitro benzoate (50 gm) and 10% palladium/carbon (5 gm) were hydrogenated under a hydrogen gas pressure of 5-6 kg for 4 hours. The reaction mass was filtered to remove catalyst. The filtrate was distilled off to remove solvent. The residue obtained was stirred in n-hexane (100 ml) at 0-5° C. The solid obtained was filtered and washed with n-hexane (25 ml) to yield the title compound (40 gm, 93% yield).
EXAMPLE 11 Preparation of 6-hydroxy-7-methoxy-quinazoline-4-one (formula XI)Methyl-2-amino-5-hydroxy-4-methoxy benzoate (50 gm), methanol (400 ml) and formamidine acetate (30 gm) were refluxed for 10 hours. The reaction mass was gradually cooled to 5-10° C. and stirred for 1 hour. The solid obtained was filtered and washed with methanol (150 ml) and dried at 50-55° C. to yield the title compound (45 gm, 92% yield).
EXAMPLE 2 Preparation of 6-(3-morpholino propoxy)-7-methoxy-4-chloroquinazoline (Formula VII)DMF (3 lt), 6-(3-chloropropoxy)-7-methoxy-4-chloro quinazoline (200 gm) and morpholine (210 gm), were heated to 70-75° C. for 6-8 hours. The reaction mass was cooled to room temperature, and methylene chloride (2.5 lt) and water (2.5 lt) were charged. The layers separated and the aqueous layer extracted with methylene chloride twice (500 ml). The combined methylene chloride layer was washed with water, dried over sodium sulphate (10 gm) and concentrated completely at 35-40° C. to yield the title compound (200 gm, 85% yield).
EXAMPLE 3 Preparation of 6-(3-chloropropoxy)-7-methoxy-4-chloroquinazoline (Formula VI)6-(3-chloropropoxy)-7-methoxyquinazoline-4-one (400 gm), thionyl chloride (3.2 lt) and DMF (100 ml) were refluxed for 7-8 hours. Thionyl chloride was distilled off completely under reduced pressure below 45° C. Methylene chloride (2.5 lt) and water (1.5 lt) were charged, stirred for 30 minutes at room temperature and the layers separated. The aqueous layer was extracted twice with methylene chloride (500 ml), the combined methylene chloride layer was washed with 1% sodium bicarbonate solution (1 lt), dried over sodium sulphate (20 gm) and concentrated under reduced pressure at 35-40° C. The residue was stirred with isopropyl alcohol (400 ml) at 40-45° C. for 1 hour, cooled to 0-5° C., the solids filtered, washed with chilled isopropyl alcohol (200 ml) and dried under vacuum at 45° C. to yield the title compound (406 gm, 95% yield).
EXAMPLE 4 Preparation of 6-(3-chloropropoxy)-7-methoxyquinazoline-4-one (Formula V)2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid (450 gm), formamide (2250 ml) and ammonium formate (200 gm) were heated to 170-180° C. for 3-4 hours. The reaction mass was concentrated under reduced pressure at 140-150° C. The residue was stirred in methanol (1000 ml) at 45-50° C. and cooled to 5-10° C. The solid obtained was filtered to yield the title compound (420 gm, 90% yield).
EXAMPLE 5 Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid (Formula IV) a) Preparation of 3-(3-chloropropoxy)-4-methoxy-6-nitrobenzoic acidMethanol (4 lt), 3-(3-chloropropoxy)-4-methoxy-6-nitro benzaldehyde (560 gm) and 30% methanolic NaOH solution (5 ml) were heated to 45° C. To this reaction mass 35% of H2O2 solution (1200 ml) was added drop wise in 3-4 hours maintaining a pH of 10.5-11.5 with 30% methanolic NaOH solution. The reaction mass was quenched into ice water (10 kg) and the pH adjusted to 2.0-3.0 using hydrochloric acid. The solid obtained was filtered, washed with 50% aqueous methanol (500 ml) and dried at 45-50° C. to yield the title compound (510 gm, 86% yield).
bi) Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid—Using Hydrogen GasEthyl acetate (3 lt), Pd/C (50 gm) and 3-(3-chloropropoxy)-4-methoxy-6-nitrobenzoic acid (500 gm) were hydrogenated under a hydrogen pressure of 5-6 kg at 35-40° C. for 3-4 hours. The reaction mass was filtered and the clear filtrate was distilled under reduced pressure at 45-50° C. To the residue, hexane (1 lt) was charged, stirred at room temperature, the solids filtered and dried at 45-50° C. to yield the title compound (403 gm, 90% yield).
(bii) Preparation of 2-amino-4methoxy-5-(3-chloropropoxy)benzoic acid—Using Hydrazine Hydrate3-(3-chloropropoxy)-4-methoxy-6-nitrobenzoic acid (100 gm), hydrazine hydrate (50 gms), neutral alumina (20 gms), charcoal (10 gms), water (50 ml) and methanol (500 ml) were mixed together. The reaction mass was heated to 50° C. A solution of ferric chloride (2 gms, 0.012M) in 50 ml methanol was introduced slowly at 55-60° C. The reaction mass was filtered over hyflo and the clear filtrate evaporated. The residue obtained was dissolved in 1.0-lit ethyl acetate, washed organic extract with water, evaporated to obtain title compound. (75 gms, 83.6%)
(biii) Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid—Using Ammonium Formate3-(3-chloropropoxy)-4-methoxy-6-nitro benzoic acid (165 gms), 5% Paladium on carbon (16.5 gms) and DMF (0.66 lit) were mixed together. The reaction mass was heated to 40° C. Ammonium formate (82.5 gms) was charged in lots maintaining temperature below 50° C. The temperature of reaction mass slowly raised to 70° C. and maintained for 2 hours. The reaction mass was cooled to 30° C. and catalyst was removed by filtration and the clear filtrate evaporated. The residue was dissolved in ethyl acetate (0.825 lit), washed with water and evaporated to yield the title compound. (125 gms, 84.5%)
EXAMPLE 6 Preparation of 3-(3-chloropropoxy)-4-methoxy-6-nitro benzaldehyde (Formula III)5-nitro isovanillin (500 gm), acetonitrile (3.5 lts), K2CO3 (750 gm) and chlorobromopropane (780 gm) were refluxed for 4 hours. The reaction mass was filtered hot, washed with acetonitrile (1 lt) and the filtrate was distilled off to remove solvent. The residue was dissolved in methylene chloride (4 lt) and washed with water. Water (3 lt) was charged to the methylene chloride layer, the pH adjusted to 7.0 to 7.5 with acetic acid, the methylene chloride layer separated, dried over sodium sulphate (50 gm) and distilled out completely under reduced pressure below 40° C. The residue was stirred with 2 volumes of n-Hexane at 40-45° C., cooled slowly to 0-5° C., the solids filtered, washed with n-Hexane (250 ml) and dried at 40-45° C. to yield the title compound (638 gm, 92% yield).
EXAMPLE 7 Preparation of 5-nitro isovanillin (Formula II)Isovanillin (500 gm) and acetic acid (1750 ml) were cooled to −5 to 0° C. To this solution, nitric acid (750 ml) was charged slowly at −5 to 0° C. with stirring. The temperature of the reaction mass was slowly raised to 25-30° C. and maintained for 12 hours. The reaction mass was quenched into ice water (4 kg), the solids filtered and washed with water (2 lt). The solids were stirred with a 1% sodium bicarbonate solution (1 lt), filtered and dried at 45-50° C. The solid was dissolved in 6 volumes of ethyl acetate, ethyl acetate was distilled off up to half the volume and 3 volumes of n-Hexane were charged slowly at 45-50° C. The reaction mass was cooled slowly to 0-5° C., maintained for 1 hour, the solids filtered, washed with 0.5 volumes of 1:1 mixture of ethyl acetate:n-Hexane and dried at 45-50° C. to yield the title compound (423 gm, 65% yield).
EXAMPLE 8 Preparation of Methyl-2-hydroxy-3-methoxy benzoate (Formula VIII) a) Preparation of 3-hydroxy-4-methoxy benzoic acidMethanol (350 ml), isovanillin (50 gm) and 30% methanolic sodium hydroxide solution (1 ml), were heated to 45° C. To this solution, 35% hydrogen peroxide solution (107 ml) was charged slowly maintaining pH at 10.5 to 11.5 using methanolic sodium hydroxide solution over a period of 2-3 hours. The reaction mass was quenched into chilled water (1 lt) and the pH adjusted to 2-3 using hydrochloric acid. The solids were filtered, washed with 50% aqueous methanol (50 ml) and dried at 45-50° C. to yield 3-hydroxy-4-methoxy benzoic acid.
b) Preparation of Methyl-2-hydroxy-3-methoxy benzoateThe solid obtained in step a), was refluxed with 10% methanolic hydrochloric acid solution (250 ml) for 6 hours. The reaction mass was quenched into chilled water (1 lt) and repeatedly extracted with methylene chloride (250 ml). The combined methylene chloride layer was washed with water (100 ml×2) and methylene chloride distilled out completely at 35-40° C. The residue was stirred in hexane (1.50 ml), at 25-30° C. The solid obtained was filtered, washed with: hexane (25 ml) and dried at 40-45° C. to yield the title compound (50 gm, 83% yield).
EXAMPLE 9 Preparation of Methyl-5-hydroxy-4-methoxy-2-nitro benzoate (Formula IX)Methyl-2-hydroxy-3-methoxy benzoate (50 gm) and acetic acid (175 ml) were cooled to 0-5° C. To this solution, 70% nitric acid solution (75 ml) was charged slowly at 0-5° C. under stirring and the reaction mass was further stirred for 18 hours. The reaction mass was quenched into chilled water (800 ml) and extracted repeatedly with methylene chloride (400 ml). The combined methylene chloride layer was washed with water, followed by 1% potassium carbonate solution (100 ml), dried over sodium sulphate and methylene chloride distilled off completely at 35-40° C. The residue was dissolved in 10% aqueous methanol (250 ml). The filtrate was gradually cooled to 0-5° C. and maintained for 1 hour. The solid obtained was filtered, washed with 10% aqueous methanol (100 ml) and dried at 40-45° C. to yield the title compound (46 gm, 74% yield).
EXAMPLE 10 Preparation of Methyl-2-amino-5-hydroxy-4-methoxy benzoate (X)Ethyl acetate (300 ml), methyl-5-hydroxy-4-methoxy-2-nitro benzoate (50 gm) and 10% palladium/carbon (5 gm) were hydrogenated under a hydrogen gas pressure of 5-6 kg for 4 hours. The reaction mass was filtered to remove catalyst. The filtrate was distilled off to remove solvent. The residue obtained was stirred in n-hexane (100 ml) at 0-5° C. The solid obtained was filtered and washed with n-hexane (25 ml) to yield the title compound (40 gm, 93% yield).
EXAMPLE 11 Preparation of 6-hydroxy-7-methoxy-quinazoline-4-one (formula XI)Methyl-2-amino-5-hydroxy-4-methoxy benzoate (50 gm), methanol (400 ml) and formamidine acetate (30 gm) were refluxed for 10 hours. The reaction mass was gradually cooled to 5-10° C. and stirred for 1 hour. The solid obtained was filtered and washed with methanol (150 ml) and dried at 50-55° C. to yield the title compound (45 gm, 92% yield).
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6297257 | 17 Dec 1998 | 2 Oct 2001 | Zambon Group S.P.A. | Benzazine derivatives phosphodiesterase 4 inhibitors |
| EP1477481A1 | 28 Jan 2003 | 17 Nov 2004 | Ube Industries, Ltd. | Process for producing quinazolin-4-one derivative |
| IN901CHE2006A | Title not available | |||
| IN903CHE2006A | Title not available | |||
| WO1996033980A1 | 23 Apr 1996 | 31 Oct 1996 | Zeneca Limited | Quinazoline derivatives |
| WO2004024703A1 | 9 Sep 2003 | 25 Mar 2004 | Astrazeneca Ab | Process for the preparation of 4- (3’-chloro-4’-fluoroanilino) -7-methoxy-6- (3-morpholinopropoxy) quinazoline |
| WO2005023783A1 | 1 Sep 2004 | 17 Mar 2005 | Astrazeneca Ab | Process for the manufacture of gefitinib |
| WO2005070909A1 | 27 Jul 2004 | 4 Aug 2005 | Natco Pharma Limited | An improved process for the preparation of gefitinib |
| WO2008125867A2 | 16 Apr 2008 | 23 Oct 2008 | Cipla Limited | Process for the preparation of gefitinib |
/////////Gefitinib, US 8350029, CIPLA