
Synthesis of (S)-pregabalin, Teva
TEVA PHARMACEUTICALS INTERNATIONAL GMBH [CH/CH]; Schusselstrasse 12 8645 Jona (CH) (For All Designated States Except US).
JANAGANI, Satyanarayana [US/US]; (US) (US only)
JANAGANI, Satyanarayana [US/US]; (US) (US only)
Improved process for preparing (S)-pregabalin, useful for treating pain, seizures, convulsions and anxiety. Also claims novel intermediates of (S)-pregabalin and their preparation method.
Pregabalin, a GABA alpha-2-delta subunit agonist, had been developed and launched by Pfizer.
Teva received a FDA approval for its generic pregabalin capsules (25, 50, 75, 100, 150, 200, 225 and 300 mg).
S)-Pregabalin, (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid, a compound having the chemical structure,

is also known as pregabalin, γ-amino butyric acid or (S)-3-isobutyl GABA. (S)-Pregabalin, marketed under the name LYRICA®, has been found to activate GAD (L-glutamic acid decarboxylase). (S)-Pregabalin has a dose dependent protective effect on seizure, and is a CNS-active compound. (S)-Pregabalin is useful in anticonvulsant therapy, due to its activation of GAD, promoting the production of GABA, one of the brain's major inhibitory neurotransmitters, which is released at 30 percent of the brains synapses. (S)-Pregabalin has analgesic, anticonvulsant, and anxiolytic activity.
Several processes for the synthesis of (S)-Pregabalin are known. For example, U.S. Patent No. 5,599,973 ("'973 patent") discloses the preparation of (S)-Pregabalin using a stoichiometric amount of (4R,5S)-(+) 4-methyl-5-phenyl-2-oxazolidinone as a chiral auxiliary that may be recycled. See, e.g., '973 patent, col. 14, 1. 29 to col. 18, 1. 23 (example 1). In general, however, the route disclosed in the '973 patent is of limited use on an industrial scale, principally due to the low temperature required for the reaction (e.g., -78°C), the use of pyrophoric reagent (e.g., butyl lithium), and a low overall yield (e.g. , 59%, 65%).
U.S. Publication No. 2003/0212290 ("'290 publication") discloses the synthesis of (S)-Pregabalin by an asymmetric hydrogenation of a cyano-substituted olefin of formula 7, to produce a cyano precursor of (S)-3-(aminomethyl)-5-methyl hexanoic acid of formula 8, which i btain (S)-Pregabalin, as described in the following scheme.
[(R,R)-MeD PHOS]Rh(COD)+BF4-

However, the disclosed method requires the use of carbon monoxide under high pressure, raising serious problems in adapting this process for production scale.
Another process is disclosed by G.M. Sammis, et al, J. Am. Chem. Soc , 125(15): 4442-43 (2003), in which an aluminum salen catalyst is used in the conjugate addition of hydrogen cyanide to a, β-unsaturated imides.

Pregabalin
This process is also not practical for large scale production due to the use of highly poisonous reagents. In addition, the last reduction step requires high hydrogen pressure, which only adds to the difficulties required for adapting this process for use on an industrial scale.
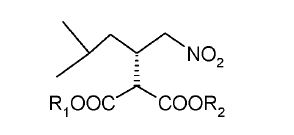
International Publication WO 2006/110783 reports several processes for preparing (S)-Pregabalin via the following intermediate and its analogues.
R^OC "COOR2
wherein Ri and R2 are independently H, a straight or branched Ci-10 alkyl, C6-10 aryl, or C3-6 allyl.
U.S. Publication Nos. 2007/0191636 and 2007/0197827 also disclose processes for preparing (S)-Pregabalin.
Thus, there is a need in the art for additional process for the preparation of (S)-Pregabalin that provide (S)-Pregabalin in high quality and high yield, and that can be adapted to large (industrial) scale production.
EXAMPLES
Example 1: Preparation of (3S)-5-methyl-3-(2-oxo-2{[(lS)-l-phenylethyllamino} ethyl) hexanoic acid (III, wherein Ar = phenyl and R = methyl) with recycling of compound (Ilia)
A. 3-isobutylglutaric acid (700g) and acetic anhydride (420g) were heated to 130-140°C and maintained for about 3 hrs. At the end of the reaction, the reaction mixture was cooled to 70-80°C and acetic acid and acetic anhydride were distilled off under vacuum. Toluene (700 mL) was added to the reaction mixture and further evaporated=for 1.5-2 hrs at 90-95°C. Another 700mL of toluene were added and the resulting 4-isobutylglutaric anhydride (IBG anhydride) solution was cooled to 25-30°C.
B. A different reactor was charged with toluene (4L), S-phenylethylamine (1.05 mol equivalent) and 4-dimethylaminopyridine (DMAP) (4.5g) and the mixture was cooled to
-25 to -15°C. The IBG anhydride solution was added and stirred at -25 to -15°C for 2-3 hrs. The mixture was heated to 25-30°C, 180 mL of aq. HC1 (30%) and water (180 mL) were added and the mixture was heated to 70-75°C. The phases were separated and the organic phase was cooled to 15-30°C and stirred for 2-2.5 hrs. The mixture was filtered and washed twice with toluene (2 vol.).
C. The toluene mother liquor, contained 226 g of the compound of formula Ilia (Ar = phenyl and R = methyl) (ee 76.7 %). The toluene was distilled off to 3 vol and 136 g acetylchloride were added. The mixture was heated to 78-82°C and stirred for 5-6 hrs. At the end of the reaction time, 1130 mL water was added at 50-60°C and the phases were separated. 47.39 g NaOH in 474 mL of water were added to the organic phase and the reaction mixture was heated to 78-82°C and stirred for 8-10 hrs. Then, the reaction mixture was cooled to 25-30°C and the pH was adjusted to 1-3 with 30% HC1. Toluene (8 vol.) was added to the mixture and the phases were separated at 80°C. The organic phase was cooled to 25-30°C and filtered. The filtrate was washed with toluene (2 vol.) and re-crystallized from toluene. Yield 44.94%, purity 97.5%, ee 99.88%.
Example 2: Preparation of (3S)-5-methyl-3-(2-oxo-2{[(lS)-l-phenylethyllamino} ethyl) hexanoic acid
A three-necked flask equipped with an addition funnel, thermometer pocket, drying tube and a mechanical stirrer, was charged with toluene (400 ml), (S)-(-)-phenylethylamine (142.35 g,1.1764 mole), and 4-dimethylaminopyridine (0.7176 g, 0.0059 mole). The mixture was cooled to a temperature of -10°C to -15°C, followed by addition of a solution of 3- isobutyl glutaric anhydride (100 g, 0.59 mole) [e.g. obtained in accordance with the process disclosed Drugs of the Future, 24 (8), 862-870 (1999) or according to Example 1 step (A) above] in toluene (100 ml), over a period of 45-60 minutes, and stirring for additional 1.5-2 hours, at a temperature of -10°C to -15°C. The mixture was then extracted with 10% aqueous solution of NaOH (500 ml), and the aqueous phase was washed with toluene (1x250 ml). The pH of the aqueous phase was adjusted to 2-2.5 by adding a solution of hydrochloric acid (1-12N). The aqueous phase was further extracted with toluene (lx 800 ml) at a temperature of 70-80°C. The toluene layer was washed with 10% sodium chloride solution {700ml) at a temperature of 70-80°C followed by crystallization to get 125 g (73.0% yield) of a white solid of (3S)-5-methyl-3-(2-oxo-2-{[(l S)-l-phenylethyl]amino}ethyl) hexanoic acid with an optical purity of 99.75 %, as measured by chiral HPLC.
The toluene mother liquor obtained from the crystallization, which contains a mixture of diastereomers [i.e. compound (Ilia) and (III) wherein Ar = phenyl and R = methyl) is then further processed in accordance with Example 1, step C, in order to convert the compound of formula (Ilia) to (III).
Example 3; Preparation of (3S)-5-methyl-3-(2-oxo-2{[(l S)-l-phenylethyllamino} ethyl) hexanoic acid

Desired major
To a cooled (0 °C) solution of 4-Isobutylglutaric anhydride (0.1 moles) in toluene is added (lS)-l-phenylethanamine (0.1 moles) slowly during 30 minutes and the mixture is warmed to 70 °C, washed with dilute HC1 followed by brine and cooled to ambient temperature during several hours. The precipitate is filtered, washed with toluene and vacuum dried until constant weight to yield (3S)-5-methyl-3-[2-oxo-2-[[(lS)-l-phenylethyl] amino] ethyl]hexanoic acid. Diastereomeric purity by HPLC = 99.5%.
The toluene mother liquor obtained from the precipitation, which contains a mixture of diastereomers [i.e. compound (Ilia) and (III) wherein Ar = phenyl and R = methyl) is then further processed in accordance with Example 1, step C, in order to convert the compound of formula (Ilia) to (III).
Example 4: Preparation of {(S)-4-methyl-2-[((S)-l-phenylethylcarbamoyl)-methyllpentvUcarbamic acid methyl ester
A three-necked flask equipped with an addition funnel, thermometer pocket, drying tube and a mechanical stirrer, was charged with acetone (25 ml), (3S)-5-methyl-3-(2-oxo-2{[(l S)-l-phenylethyl]amino} ethyl) hexanoic acid (5 g, 0.0172 mole), and with
triethylamine (2.17g, 0.0215 mole), and cooled to -10° to -20°C followed by addition of solution of ethyl chloroformate (2.05 g, 0.0189 mole in 5 ml acetone). The mixture was stirred for 1 hour at a temperature of -10° to -20°C, followed by addition of solution of sodium azide (2.8g, 0.0429 mole in water). The resulted slurry was maintained for 1 hour at -10° to -20°C, quenched over ice water followed by extracting the mass with sufficient amount of toluene. The toluene layer was slowly added over a refluxing mixture of toluene and methyl alcohol, followed by stirring for 2 to 4 hours. The stripping off the solvent results in 4.95 g (89.7% yield) of {(S)-4-methyl-2-[((S)-l-phenylethylcarbamoyl)-methyl]pentylcarbamic acid methyl ester (120) with a purity of 97.4% area, as measured by HPLC.
Example 5: Preparation of (S)-Pregabalin
A 0.2 1 reactor was loaded with 70% sulfuric acid (200 g) containing compound 26 (10 g, 0.031 mole), and was heated to 115-120°C for 5-10 hours, and then cooled to room temperature, i.e., about 20° to about 25°C. An aqueous 40% sodium hydroxide solution was added in an amount sufficient to provide a pH of 1. The solution was then extracted with 35 ml of iso-butanol, the organic layer was separated, and Β¾Ν was added in an amount sufficient to provide a pH of 4. The (S)-Pregabalin was precipitated, filtered, and washed with 10 ml of iso-butanol. After drying at 55°C under vacuum, (S)-Pregabalin was obtained as white crystals in a 40.4% yield. Purity: 99.95% area by HPLC.
Example 6: Preparation of (S)-Pregabalin
A flask was loaded with 47% HBr (12 ml), water (6 ml), and compound 26 (6 g), and then was heated to reflux for 3 hours. The solution was cooled to room temperature, and water (12 ml) was added. An aqueous 47% sodium hydroxide solution was added to obtain
pH of 3. The solution was then extracted twice with isobutanol (15 ml), the combined organic layers were evaporated and fresh isobutanol was added (15 ml). B¾N (3.8 g) was added. The mixture was cooled to 2°C for 1 hour, then (S)-Pregabalin was filtered, and washed with of iso-butanol (3 ml). After drying at 55°C under vacuum, (S)-Pregabalin was obtained as white crystals in a 90% yield.
Example 7: Conversion of the Compound of Formula 4 to (S)-Pregabalin: Example 14 of International Publication No. WO 2007/035890
A 0.2 1 reactor was loaded with 70% sulfuric acid (200 g) containing compound 26 (10 g, 0.031 mole), and was heated to 115-120°C for 5-10 hours, and then cooled to room temperature, i.e., about 20° to about 25°C. An aqueous 40% sodium hydroxide solution was added in an amount sufficient to provide a pH of 1. The solution was then extracted with 35 ml of iso-butanol, the organic layer was separated, and Bu3N was added in an amount sufficient to provide a pH of 4. The (S) Pregabalin was precipitated, filtered, and washed with 10 ml of iso-butanol. After drying at 55°C under vacuum, (S)-Pregabalin was obtained as white crystals in a 40.4% yield. Purity: 99.95% area by HPLC.
Compound 26 has the following chemical structure:
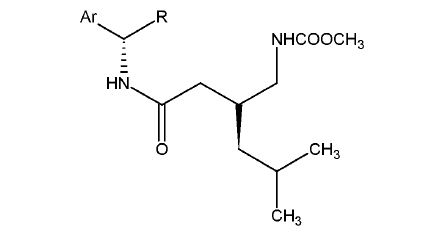
wherein Ar is a C6-1o aromatic group, and R is a straight or branched C1-4 alkyl, ester or carboxylic acid.
Example 8: Conversion of the Compound of Formula 4 to (S)-Pregabalin: Example 16 of International Publication No. WO 2007/035890
A flask was loaded with 47% HBr (12 ml), water (6 ml), and compound 26 (6 g), and then was heated to reflux for 3 hours. The solution was cooled to room temperature, and water (12 ml) was added. An aqueous 47% sodium hydroxide solution was added to obtain pH of 3. The solution was then extracted twice with isobutanol (15 ml), the combined organic layers were evaporated and fresh isobutanol was added (15 ml). Bu3N (3.8 g) was added. The mixture was cooled to 2°C for 1 hour, then (S)-Pregabalin was filtered, and washed with of iso-butanol (3 ml). After drying at 55°C under vacuum, (S)-Pregabalin was obtained as white crystals in a 90% yield.
/////////////// (S)-pregabalin, WO 2017019791