

paroxetine
PROCESS FOR THE PREPARATION OF PAROXETINE
This invention relates to processes for the manufacture of paroxetine and pharmaceutically acceptable salts thereof which are suitable for large scale commercial operation.
Pharmaceutical products with antidepressant and anti-Parkinson properties are described in US-A-3912743 and US-A-4007196. An especially important compound among those disclosed is paroxetine, the (-)trans isomer of 4-(4 -fluorophenyl)-3-(3',4 - methylenedioxy-phenoxymethyl)-piperidine. This compound is used in therapy as the hydrochloride salt to treat ter alia depression, obsessive compulsive disorder (OCD) and panic.
Various processes have been described for the preparation of paroxetine, for example in US 4,007,196, EP 0219,934, EP 0223, 334, EP 223, 403, EP 0300,617 and Acta Chemica Scandinavica (1996) volume 50 page 164. A particularly useful starting material employed in processes described therein is the alkaloid arecoline (1)

In these processes, arecoline is used to prepare piperidines using a literature procedure (J.T. Plati, A.K Ingerman and W Wenner, Journal of Organic Chemistry (1957) Volume 22 pages 261-265). Plati et al describe the reaction of arecoline with phenyl magnesium bromide in diethyl ether to prepare l-methyl-3-carbomethoxy-4-phenyl piperidine. Thus in the process described in US-A-4007196, arecoline base is liberated from the hydrobromide salt and reacted with the Grignard reagent 4-fluorophenyl magnesium bromide using the procedure of Plati et al to give a piperidine ester of structure (2). This piperidine ester is converted to a piperidine carbinol of structure (3), which is coupled with sesamol, then deprotected. to give paroxetine (4).

Paroxetine is the (-) trans isomer of 4-(4'-fluorophenyl)-3-(3',4'-methylenedioxy- phenoxymethyl)-piperidine. The above described processes produce compounds of structure (2) as a mixture of enantiomers, and conversion of compounds of structure (2) to useful pharmaceuticals will normally require a resolution stage. Particularly useful forms of compounds (2) and (3) are thus compounds (A) and (B) which are in the (-) trans configuration:

(A) (B)
The prior art processes for the_ preparation of paroxetine (4) from arecoline (1) suffer from a number of disadvantages which render them unsuitable for large scale manufacture. In particular, the reaction of arecoline with the Grignard reagent is carried out in diethyl ether, and the resulting piperidine ester is extracted into ether and subsequently distilled under vacuum. These conditions are hazardous and unsuitable for lar^e scale manufacture of compounds of structure (2). We have also have found that the Plati procedure generates a thick unstirrable gel. and that this gel is not broken down by adding further diethyl ether.
Furthermore we have found that other ether solvents conventionally used in Grignard reactions, such as tetrahydrofuran or diisopropyl ether result in little if any of the desired 1 ,4-conjugate addition product, as the major product arises from attack of the Grignard reagent on the ester grouping (so called 1,2- addition).
We have discovered that the Plati et al procedure can be rendered suitable for large scale manufacture by varying the conditions under which the Grignard reagent is used, enabling the stirring problems to be overcome and the use of diethyl ether eliminated or significantly reduced.
In an unpublished patent application we disclose a process for the preparation of a compound of structure (5)

(5)
in which R and R' are independently an alkyl, aryl, or arylalkyl group, most suitably lower alkyl, which comprises reacting a compound of structure (6) where R and R' are as defined for structure (5)

The Grignard reagent may either be prepared in the chosen reaction solvent, or prepared in an ether solvent and the ether subsequently removed by distillation and replaced by the chosen solvent. When little or no additional solvent is employed, the Grignard reagent may be partially or completely insoluble, but the resulting reaction suspension is stirrable and compatible with large scale operation. When a significant proportion of suitable additional solvent is employed, a completely clear reaction solution may be obtained, rendering the process particularly suitable for industrial scale operation.
Surprisingly we have found that by using the above-described processes the reaction is more efficient, and the large excess of Grignard reagent specified by Plati (2 molar equivalents) can be significantly reduced without loss in yield. We have also found that the reaction is equally efficient if the order of addition of the reagents is reversed, i.e. the Grignard reagent is added to the tetrahydropyridine ester. A further advantage we have found is that the crude product is sufficiently pure to be carried forward to the next synthetic step without the need for the distillation specified by Plati.
Compounds of formula (6) may be prepared from the natural products guvacine, arecaidine or arecoline, by conventional methods, or by synthesis from other materials. A particularly convenient synthetic procedure involves the esterification, quaternisation and partial reduction of nicotinic acid [see for example Journal of Organic Chemistry (1955), volume 20, pages 1761-1765; Journal of Chemical Research (1983), volume 10, pages 2326 - 2342; Journal of Pharmaceutical Sciences (1992), volume 81, pages 1015 -1019; and references quoted therein].

Other methods for the preparation of compounds of structure (6) are given in Tetrahedron (1989) volume 45 pages 239-258, and Heterocycles ( 1990) volume 30 pages 885 - 896.
In a further unpublished patent application we propose an alternative procedure based on the finding that a mixture of crystalline arecoline hydrobromide in a suitable organic solvent may treated with a suitable anhydrous strongly basic reagent to generate a solution which may be reacted directly with the Grignard reagent. This process is higher yielding than the prior art process, has fewer processing steps, and exposure of operators to the toxic arecoline base is avoided. By comparison with arecoline base, the hazards associated with handling arecoline hydrobromide, which is a non-volatile crystalline solid and freely soluble in water, are greatly reduced. The process of this invention is therefore particularly suitable for large scale manufacture.
Hereinafter we describe processes for the preparation of paroxetine starting from arecoline or an arecoline analogue which are suitable for large scale manufacture. These processes proceed through an intermediate of formula (5) above, which is advantageously made by the above described process rather than the previously known process of Plati.
A procedure describing the conversion of a piperidine ester of structure (2) to paroxetine is described in US-A-4007196 whereby the initially formed piperidine methyl ester prepared by the Plati method is epimerised to the trans form, hydrolysed to the piperidine acid, converted to the acid chloride and reacted with (-) menthol to give the corresponding menthol ester. This menthol ester is distilled, converted to the hydrobromide salt and fractionally crystallised to give a single enantiomer. The resolved menthol ester is liberated from the salt and reduced with lithium aluminium hydride to give the (-) trans carbinol, compound (B), which is coupled with sesamol, then deprotected to give paroxetine. This multi-step process, while sufficient for investigational purposes, is not suitable for large scale manufacture.
A shorter procedure is described in the form of a flow chart in Acta Chemica Scandinavica (1996) volume 50 page 164, but few details of the conditions are supplied. The first part of this procedure is represented in Scheme ( 1), the (-) trans carbinol of structure (B) being converted to paroxetine by the further steps of coupling to sesamol, and deprotecting.
Scheme 1

A procedure for carrying out Step 1 of Scheme 1 is described by J.T. Plati, A.K Ingerman and W Wenner, Journal of Organic Chemistry (1957) Volume 22 pages 261-265.
5 Procedures for carrying out Step 2 of Scheme 1 are described in Example 1 of US-A- 4007196 and Example 2 of EP 0219,934 A procedure for carrying out Step 3 of Scheme 1 is outlined on page 3 of EP 0219,934
Procedures for carrying out Step 4 of Scheme 1 are described in Examples 5 and 8 of EP 0223,334
http://www.google.com/patents/WO2001029032A1?cl=en
We have also found that (-) trans-4-(4'-fluorophenyl)-3-hydroxymethyl-l- methylpiperidine (B) may be prepared from arecoline by an alternative sequence of steps involving the formation and reduction of (-) trans l-methyl-3-carbomethoxy-4-(4'- fluorophenyl) piperidine (A), and employed in the synthesis of paroxetine. Such a process is outlined in Scheme 2

(-) trans carbinol (-) trans ester
A procedure for carrying out Step 1 of Scheme 2 is described by J.T. Plati, A.K Ingerman and W Wenner, Journal of Organic Chemistry (1957) Volume 22 pages 261-265.
Procedures for carrying out Step 2 of Scheme 2 are described in Example 1 of US-A- 4007196 and Example 2 of EP 0219,934
An outline method for the chemical resolution of trans l-methyl-3-carbomethoxy-4-(4'- fluorophenyl) piperidine (Step 3 of Scheme 2) using unspecified optical forms of mandelic acid or dibenzoyl tartaric acid has been described in the literature in the form of a flowchart [Acta Chemica Scandinavica (1996) volume 50 page 164], but no details of the conditions are given. The same flowchart outlines the reduction of the (-) trans ester (A) to the (-) trans carbinol (B), step 4 of Scheme 2.
We have made numerous attempts carry out this resolution procedure but have been unable to obtain any crystallise salts using either mandelic acid or dibenzoyl tartaric acid in a wide range of organic solvents. In addition, no chemical or physical properties for the individual (+) and (-) optical isomers of trans l-methyl-3-carbomethoxy-4-(4'- fluorophenyl) piperidine or analogous trans compounds of structure (5) have been reported, either as salts or in the free base form.
We therefore conclude that no workable process for obtaining (-) trans l-methyl-3- carbomethoxy-4-(4'-fluorophenyl) piperidine (A) or analogous resolved trans compounds of structure (5) is available in the prior art.
We have surprisingly found that the desired (-) form of a trans ester of structure (5) can be prepared by enzymatic resolution of a racemic trans ester of structure (5), enabling paroxetine to be manufactured from arecoline by the steps outlined in Scheme 2.
Accordingly in a second aspect of this invention we provide a process for the large scale manufacture of paroxetine and pharmaceutically acceptable salts thereof from an arecoline derivative and a 4-fluorophenylmagnesium halide which comprises the steps
a) reacting a salt of an arecoline derivative of formula (6)
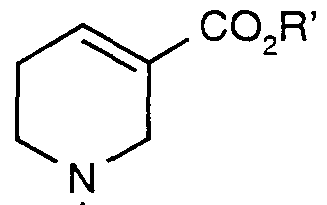
R (6) with a 4-fluorophenylmagnesium halide and extracting the cis/trans piperidine ester of formula (5)

(5)
b) converting cis/trans piperidine ester of formula (5) to the corresponding trans ester by epimerising with a strong base,
c) enzymatic resolution of the trans piperidine ester to give the (-) trans piperidine ester,
d) reducing the (-) trans piperidine ester with a hydride reducing agent to obtain the corresponding (-) trans carbinol,
e) forming a sulphonate ester of the (-) trans carbinol then coupling with sesamol, to obtain an N-protected paroxetine,
f) deprotecting the N-protected paroxetine via formation of a carbamate, followed by hydrolysis to generate paroxetine base,
g) isolating the paroxetine base or forming a paroxetine salt by contacting the paroxetine base with a source of a preferably pharmaceutically acceptable acid, and isolating the salt.
More specifically, a particular embodiment of this aspect of the invention comprises a) reacting an arecoline salt with a 4-fluorophenylmagnesium halide, optionally isolating the intermediate arecoline base, extracting and optionally isolating cis/trans l-methyl-3- carbomethoxy-4-(4 -fluorophenyl) piperidine,
b) epimerising the cis/trans l-methyl-3-carbomethoxy-4-(4 '-fluorophenyl) piperidine to the corresponding trans ester with a strong base, with optional isolation of the trans ester.
c) enzymatic resolution of the trans ester to give the (-) trans ester, liberating, extracting and optionally isolating the (-) trans ester,
d) reducing (-) trans l-methyl-3-carbomethoxy-4-(4 -fluorophenyl) piperidine with a hydride reducing agent and optionally isolating the (-) trans carbinol, that is (-) trans-4- (4'-fluorophenyl)-3-hydroxymethyl- 1 -methylpiperidine,
e) forming and optionally isolating a sulphonate ester of the (-) trans carbinol then coupling with sesamol, and optionally isolating the resulting N-protected paroxetine,
f) deprotecting the N-protected paroxetine via formation and optional isolation of a carbamate, followed by a hydrolysis reaction, generating and optionally isolating paroxetine base,
g) forming a paroxetine salt by contacting the paroxetine base with a source of a pharmaceutically acceptable acid, optionally converting to a second paroxetine salt, and isolating drying and optionally recrystallising the final product.
Preferably two or more of the steps are carried out in a common reaction solvent, optionally with one or more additional solvents, and optionally combining one or more of the steps a) to g).
The compound of formula (6)js most conveniently arecoline. Suitable arecoline salts at step a) are the hydrobromide and hydrochloride. A preferred arecoline salt is the hydrobromide.
Suitable 4-fluorophenylmagnesium halides at step a) are 4-fluorophenylmagnesium bromide and 4-fluorophenylmagnesium chloride. A preferred halide is the bromide. Suitable strong bases at step b) include sodium methoxide, sodium ethoxide and potassium tert-butoxide. A preferred strong base is sodium methoxide.
At step c), the chosen enzyme may selectively hydrolyse the unwanted (+) trans isomer to the corresponding acid, which may be removed by a conventional extraction, for example with an aqueous base, leaving the desired (-) trans isomer as the ester for further processing.
Alternatively, the chosen enzyme may selectively hydrolyse the desired (-) trans isomer of the ester to the corresponding (-) trans acid, compound (C)

(C) which is recovered by extraction with an aqueous base, and re-esterified to give the (-) trans ester. The (+) trans ester is unaffected by the enzyme treatment and may be recovered from the organic phase of this extraction.
In a particularly useful alternative aspect, the (-) trans acid of formula (C) is reduced directly to the desired (-) trans carbinol, for example with a borohydride reducing agent, thus avoiding the re-esterification step.
Example 12 Preparation of (-) trans 4-(4'-f_uorophenyl)-3-(3',4'-methylenedioxy - phenoxymethyl) piperidine (paroxetine free base).
Powdered potassium hydroxide (3.0 g) is added to a solution of (-) trans 4-(4'- fluorophenyl)-3-(3',4'-methylenedioxyphenoxymethyl)- 1 -phenoxycarbonyl piperidine (3.6g) in toluene (100 ml) and the well stirred mixture is refluxed for 2 hours. The mixture is cooled to ambient temperature, treated with water (100 ml), stirred well and the phases separated. The toluene phase is washed with water (50 ml), and partially evaporated at atmospheric or reduced pressure to give an anhydrous toluene solution of paroxetine free base. If a solvent-free product is desired, the toluene solution may be further distilled at atmospheric or reduced pressure until no more solvent can be removed, to give paroxetine free base as an oil.
A yield of about 2.5 g is obtained
Example 13
Preparation of paroxetine methane sulphonate
A toluene solution ( 1.0 L) containing unpurified paroxetine base (approximately 225 g) is charged to a nitrogen purged reactor and stirred at 20°C. The vessel is seeded with paroxetine methanesulfonate, then a solution of methane sulfonic acid (70 g) in propan-2- ol (0.4L) is added slowly over a period of 50 minutes. Paroxetine methansulfonate is precipitated as a white crystalline solid during the addition, and the temperature at the end of the addition rises to about 30°C. The suspension is stirred for a further 1 hour, during which time the temperature is reduced to 22°C. The product is collected by filtration, washed on the filter with propan-2-ol (2 x 0.4 L) and dried in a vacuum oven at 40°C for 24 hours.
A yield of about 230 g is obtained

Description
A potent selective 5-hydroxytryptamine reuptake inhibitor used as a treatment of major depression.
| |||||||||||||||||||||||
| Specifications | |||||||||||||||||||||||
| |||||||||||||||||||||||
Example 14
Preparation of paroxetine hydrochloride hemihydrate.
A solution of paroxetine free base ( 13.5 g) in toluene (300 ml) is stirred at room temperature and concentrated hydrochloric acid (5.2 ml) is added. The mixture is stirred for 2 hours, then the product is collected, washed with a 1: 1 mixture of toluene and water (25 ml) and dried at 50 °C to give paroxetine hydrochloride hemihydrate.
The product may be recrystallised from aqueous propan-2-ol. Example 15
Preparation of paroxetine hydrochloride anhydrate Form A
i) Trans (-)-4-(4'-fluorophenyl)-3-(3 ',4'-methylenedioxyphenoxymethyl)-N- phenoxycarbonyl piperidine (25 g) and potassium hydroxide flake (22.5 g) are suspended in toluene (375 ml) and the reaction mixture heated to reflux under nitrogen with vigorous stirring for 3 hours. The suspension is cooled to room temperature, washed with water (250 ml), and the layers separated. The organic layer is warmed to 50°C, then concentrated hydrochloric acid (6 ml) is added and the reaction mixture heated to reflux. Approximately half the toluene is removed by distillation to give an anhydrous toluene solution of paroxetine hydrochloride.
The cooled toluene solution is diluted with acetone (300 ml), and the crystalline paroxetine hydrochloride acetone solvate is collected, washed with acetone and dried in vacuum.
The paroxetine hydrochloride acetone solvate is desolvated to paroxetine hydrochloride anhydrate Form A as described in WO96/24595.
ii) (-)-Trans-4-(4'-fluorophenyl)-3-(3',4'-methylenedioxyphenoxymethyl)- 1 - phenoxycarbonylpiperidine (90 g) is heated with potassium hydroxide (8.5 g) in toluene (1500 ml) at reflux for 3 hours, cooled, washed with hot water, acidified with hydrochloric acid, and distilled to approximately one quarter volume under vacuum. Hot propan-2-ol (2000 ml) is added and the mixture cooled slowly with vigorous stirring until the temperature reaches 20°C. After stirring for a further 2 hours, the product is filtered, washed with propan-2-ol, and^dried under vacuum, to give paroxetine hydrochloride propan-2-ol solvate.
The paroxetine hydrochloride propan-2-ol solvate is desolvated to paroxetine hydrochloride anhydrate Form A as described in WO96/24595.
PATENT CITATIONS
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1998002556A2 * | Jul 14, 1997 | Jan 22, 1998 | Linden Gledhill | Screening for and use of an esterase for a stereospecific resolution |
| WO1998045263A1 * | Apr 7, 1998 | Oct 15, 1998 | Gian Luca Araldi | Analogs of cocaine |
| WO2000020390A1 * | Oct 7, 1999 | Apr 13, 2000 | Univ Georgetown | Monomeric and dimeric heterocycles, and therapeutic uses thereof |
| EP0219934A1 * | Aug 6, 1986 | Apr 29, 1987 | Beecham Group Plc | Process for the preparation of aryl-piperidine esters |
| EP0223334A1 * | Aug 6, 1986 | May 27, 1987 | Beecham Group Plc | Process for the preparation of aryl-piperidine carbinols |
| EP0614986A1 * | Feb 23, 1994 | Sep 14, 1994 | Synthelabo | Enzymatic racemate cleavage of 2-pipéridine alkylcarboxylates and their use as synthesis intermediates |
| US4007196 * | Jul 23, 1975 | Feb 8, 1977 | A/S Ferrosan | 4-Phenylpiperidine compounds |
| US5948914 * | Apr 2, 1998 | Sep 7, 1999 | Sumika Fine Chemicals Co., Ltd. | Preparing 4-(4-fluorophenyl)-3-hydroxymethylpiperidine by optically resolving with o-chlorotartranilic acid; chemical intermediate for drugs |
| US2546652 | Jul 30, 1949 | Mar 27, 1951 | Hoffmann La Roche | Pyridindenes and process for their manufacture |
| US3912743 | Jan 21, 1974 | Oct 14, 1975 | Ferrosan As | 4-Phenylpiperidine compounds |
| US4007196 | Jul 23, 1975 | Feb 8, 1977 | A/S Ferrosan | 4-Phenylpiperidine compounds |
| US5672612 * | Sep 9, 1996 | Sep 30, 1997 | Pentech Pharmaceuticals, Inc. | Amorphous paroxetine composition |
| US5872132 * | Oct 18, 1996 | Feb 16, 1999 | Smithkline Beecham Corporation | Antidepressants |
| US5962689 | Aug 19, 1997 | Oct 5, 1999 | Brantford Chemicals Inc. | Biosynthesis of antiserotonin agent |
| US6172233 | Jan 15, 1998 | Jan 9, 2001 | Smithkline Beecham Plc | Process for making paroxetine |
| US6436956 * | Nov 24, 1997 | Aug 20, 2002 | Brantford Chemicals Inc. | Less hygroscopic antidepressant |
| WO2001017966A1 | Sep 8, 2000 | Mar 15, 2001 | David Crowe | Process for the preparation of 1-methyl-3-carbomethoxy-4-(4'-fluorophenyl)-piperidine |
| WO2001029032A1 | Oct 20, 2000 | Apr 26, 2001 | David Crowe | Process for the preparation of paroxetine |
NON-PATENT CITATIONS
| Reference | ||
|---|---|---|
| 1 | * | ENGELSTOFT M ET AL: "SYNTHESIS AND 5HT MODULATING ACTIVITY OF STEREOISOMERS OF 3-PHENOXYMETHYL-4-PHENYLPIPERIDINES" ACTA CHEMICA SCANDINAVICA,DK,MUNKSGAARD, COPENHAGEN, vol. 50, no. 2, 1996, pages 164-169, XP002074608 ISSN: 0904-213X cited in the application |
additional info


see...........http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-50532010000500009

http://pubs.rsc.org/en/content/articlelanding/2010/np/b924964h#!divAbstract

org lett 2008 10 1389

http://www.google.com.ar/patents/WO2009035652A1?cl=en

The HKR of racemic anti- or syn-3-substituted epoxy esters catalyzed by a Co(III)salen complex provides ready access to the corresponding enantioenriched 3,4-disubstituted γ-butyrolactones and 3-substituted epoxy esters . This strategy has been successfully employed in the formal synthesis of biologically active 3,4-disubstituted piperidine derivatives, (−)-paroxetine and Ro 67-8867 and a natural product, (+)-eldanolide.

http://pubs.rsc.org/en/content/articlelanding/2013/ob/c3ob27321k#!divAbstract

http://www.nature.com/ncomms/2014/141118/ncomms6474/full/ncomms6474.html

http://pubs.rsc.org/en/content/articlelanding/2003/nj/b209181j#!divAbstract
79. 4-(4-Fluorophenyl)-N-alkylnipecotinate esters of general formula A represent key intermediates in the synthesis of 4-arylpiperidine-based compounds. It is noteworthy that 4-arylpiperidine is an important structural motif in many biologically active compounds (M. Engelstoft and J. B. Hansen, Acta Chemical Scandinavica, 50, 1996, pp. 164–169).

An example is (−)-menthyl (3S,4R)-trans-4-(4-fluorophenyl)-N-methylnipecotinate hydrobromide (1) which is a key intermediate in the synthesis of paroxetine. Paroxetine (Paxil®) is a highly effective chiral pharmaceutical that is useful for the treatment of depression and obsessive compulsive disorder.

The use of this compound for this purpose was disclosed in U.S. Pat. Nos. 3,912,743 and 4,007,196 whereby 4-fluorophenylmagnesium bromide was added to arecoline. The resulting adduct was epimerized and the methyl ester functionality hydrolyzed, activated using thionyl chloride, esterified using (−)-menthol, and salt formation using hydrobromic acid to provide compound 1, as depicted in Scheme 1, which was further elaborated to paroxetine using standard procedures.

The procedure disclosed in these patents for the key Grignard conjugate addition step was based on a procedure developed by Plati et al. (U.S. Pat. No. 2,546,652 and Journal of Organic Chemistry, 22, 1957, pp. 261–265) for the reaction of phenylmagnesium bromide in diethyl ether with arecoline, also in diethyl ether. Thus, a major deficiency of this process, and likewise the processes disclosed in U.S. Pat. Nos. 3,912,743 and 4,007,196, was the diethyl ether in both the arylmagnesium bromide reagent and the reaction media. Diethyl ether is a highly flammable solvent which is undesirable to use industrially. According to patents by Ward [(U.S. Pat. No. 6,172,233) and Ward et al. (WO 01/17966A1 and WO 01/29032A1)], the use of other ether solvents conventionally used in Grignard reactions, such as tetrahydrofuran (THF) or diisopropyl ether, furnished little, if any, of the desired 1,4-conjugate addition product, with the main by-product arising from 1,2-addition of the Grignard reagent on the ester grouping. From an industrial perspective, a multistep transformation in which one step resulted in, “little if any of the desired product” (for instance <10% yield) would be prohibitively expensive. Compounding this deficiency, if this process were to be used for the synthesis of paroxetine, is the fact that the low yielding step occurs at a rather late-stage in the paroxetine process, thereby necessitating the processing of large volumes of intermediary products in order to reach the Grignard reaction step. Also, disclosed in the Ward patent was the observation that when performing the reaction using the process described by Plati et al., the reaction mixture purportedly generated thick unstirrable gels.
These deficiencies were purportedly overcome by Ward by the use of a reaction solvent mixture which was non-wholly ether, as utilized by Plati et al. As well, the Ward patents purport that the use of organometallic compounds in place of the Grignard reagent also overcame these deficiencies. However, in all examples in the Ward patents, the Grignard reagent used was always a 2M solution of 4-fluorophenylmagnesium bromide in diethyl ether. Specifically, in examples 2, 3, 4 and 5 of U.S. Pat. No. 6,172,233, the weight percentage of diethyl ether introduced by the 4-fluorophenylmagnesium bromide in diethyl ether reagent relative to the total reaction volume was about 23 to 31% range. Therefore the disadvantage of having a process which necessitated diethyl ether, with all of the disadvantages associated with this solvent, largely remained. In example 1 of the same patent, the diethyl ether is removed from the 2M 4-fluorophenylmagnesium bromide reagent prior to the addition to arecoline by co-distillation with toluene. However, this requires an extra process operation and, again, does not avoid the use of diethyl ether on an industrial scale.
Similar reactions have also been utilized for transformations of this type. For instance, Murthy and Rey in U.S. Pat. No. 5,962,689 disclose the stereoselective addition of 4-fluorophenylmagnesium bromide to various 3,4-unsaturated-3-piperidine esters, amides and N-enoylsultams in toluene. Xu and Trudell (J. Heterocyclic Chem., 33, 1996, pp. 2037–2039) also described the addition of various arylmagnesium bromide reagents, including 4-fluorophenylmagnesium bromide, to R-(−)-anhydroecgonine methyl ester substrates in dichloromethane. The disadvantage in both of these publications is that a solution of the aryl Grignard reagent in diethyl ether was employed.
79. Organocatalytic asymmetric total synthesis of (R)-rolipram and formal synthesis of (3S, 4R)-
paroxetine” P. S. Hynes, P. A. Stupple, D. J. Dixon,Org. Lett. 2008, 10, 1389
http://www.google.com/patents/US7138523
http://newdrugapprovals.org/
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO