
WO 2016200930, New patent, Citarinostat, Acetylon Pharmaceuticals Inc
| citarinostat |
Acetylon Pharmaceuticals Inc
(WO2016200930) METHODS OF MAKING PROTEIN DEACETYLASE INHIBITORS

(I)
Compound (I) is disclosed in U.S. Patent No. 8,148,526 as an HDAC inhibitor.
Example 2 of U.S. Patent Application Publication No. 2015/0099744 discloses a synthesis of compound (I). As detailed herein in Example 3, this synthesis procedure resulted in the formation of significant amounts of de-chlorination and chlorine-migration side products. These impurities have solubilities that are similar to the solubilities of the desired
intermediates. Removal of the impurities is very challenging, requiring lengthy work-ups, involving numerous washes, triturations and crystallizations. Triturations, in particular, are known to be inefficient and unscalable processes. When compound (I) was prepared according to Example 2, the necessary purification steps resulted in a significant loss of desired intermdiates, led to a modest overall yield, and rendered further industrial scale up of the synthesis route unpractical. There remains a need for new methods for the synthesis of compound (I), and related compounds, that minimize the formation of impurities, and that are amenable to industrial scale-up.
BRIEF DESCRIPTION OF THE DRAWINGS
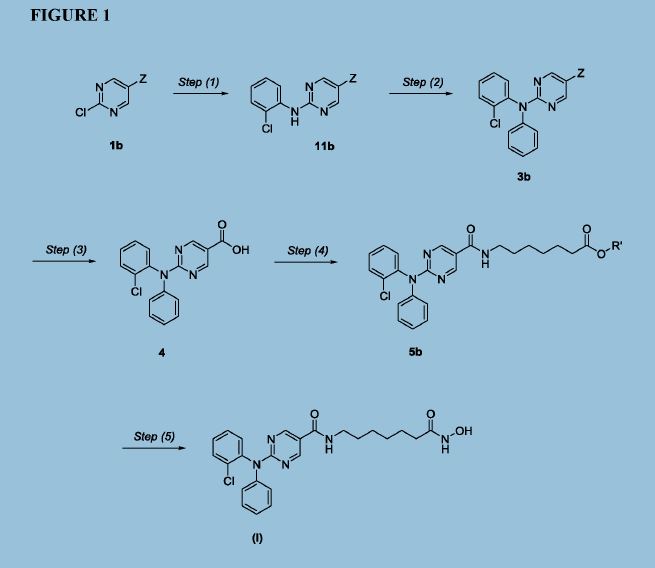
Figure 1 depicts a generic synthesis of compound (I) according to the improved method described herein.
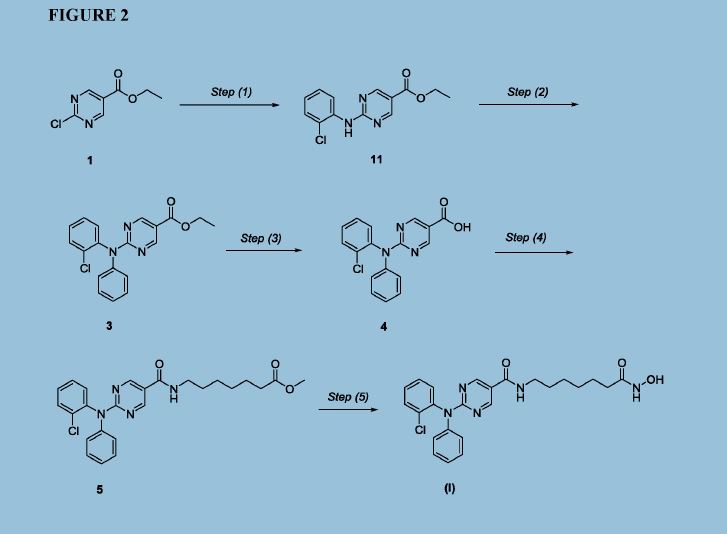
Figure 2 depicts a specific synthesis of compound (I) according to the improved method described herein.
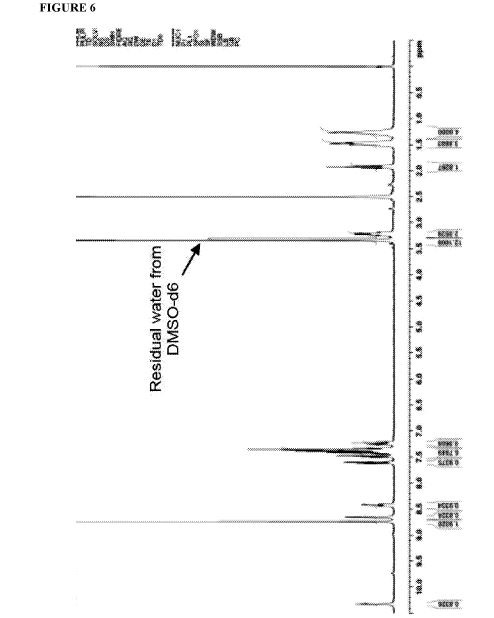
Figure 6 depicts 1HNMR data for compound (I).

Acetylon president and CEO Walter Ogier
Example 1: Comparative Synthesis of 2-(diphenylamino)-N-(7-(hydroxyamino)-7-oxoheptyl) pyrimidine-5-carboxamide
Reaction Scheme

Synthesis of Intermediate 2: A mixture of aniline (3.7 g, 40 mmol), compound 1 (7.5 g, 40 mmol), and K2C03 (11 g, 80 mmol) in DMF (100 ml) was degassed and stirred at 120 °C under N2 overnight. The reaction mixture was cooled to r.t. and diluted with EtOAc (200 ml), then washed with saturated brine (200 ml χ 3). The organic layers were separated and dried over Na2S04, evaporated to dryness and purified by silica gel chromatography (petroleum ethers/EtOAc = 10/1) to give the desired product as a white solid (6.2 g, 64 %).
Synthesis of Intermediate 3: A mixture of compound 2 (6.2 g, 25 mmol), iodobenzene (6.12 g, 30 mmol), Cul (955 mg, 5.0 mmol), Cs2C03 (16.3 g, 50 mmol) in TEOS (200 ml) was degassed and purged with nitrogen. The resulting mixture was stirred at 140 °C for 14 hrs.
After cooling to r.t., the residue was diluted with EtOAc (200 ml). 95% EtOH (200 ml) and H4F-H20 on silica gel [50g, pre-prepared by the addition of H4F (lOOg) in water (1500 ml) to silica gel (500g, 100-200 mesh)] was added, and the resulting mixture was kept at r.t. for 2 hrs. The solidified materials were filtered and washed with EtOAc. The filtrate was evaporated to dryness and the residue was purified by silica gel chromatography (petroleum ethers/EtOAc = 10/1) to give a yellow solid (3 g, 38%).
Synthesis of Intermediate 4: 2N NaOH (200 ml) was added to a solution of compound 3 (3.0 g, 9.4 mmol) in EtOH (200 ml). The mixture was stirred at 60 °C for 30min. After evaporation of the solvent, the solution was neutralized with 2N HCl to give a white precipitate. The suspension was extracted with EtOAc (2 χ 200 ml), and the organic layers were separated, washed with water (2 χ 100 ml), brine (2 χ 100 ml), and dried over Na2S04. Removal of the solvent gave a brown solid (2.5 g, 92 %).
Synthesis of Intermediate 6: A mixture of compound 4 (2.5 g, 8.58 mmol), compound 5 (2.52 g, 12.87 mmol), HATU (3.91 g, 10.30 mmol), and DIPEA (4.43 g, 34.32 mmol) was stirred at r.t. overnight. After the reaction mixture was filtered, the filtrate was evaporated to dryness and the residue was purified by silica gel chromatography (petroleum ethers/EtOAc = 2/1) to give a brown solid (2 g, 54 %).
Synthesis of 2-(diphenylamino)-N-(7-(hydroxyamino)-7-oxoheptyl)pyrimidine-5-carboxamide: A mixture of the compound 6 (2.0 g, 4.6 mmol), sodium hydroxide (2N, 20 mL) in MeOH (50 ml) and DCM (25 ml) was stirred at 0 °C for 10 min. Hydroxylamine (50%) (10 ml) was cooled to 0 °C and added to the mixture. The resulting mixture was stirred at r.t. for 20 min. After removal of the solvent, the mixture was neutralized with 1M HCl to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48%).
Example 2: Comparative Synthesis of 2-((2-chlorophenyl)(phenyl)amino)-N-(7- (hydroxyamino)-7-oxoheptyl)pyrimidine-5-carboxamide - Compound (I)
Reaction Scheme

Step (1)
Synthesis of Intermediate 2: A mixture of aniline (3.7 g, 40 mmol), ethyl 2-chloropyrimidine-5-carboxylate 1 (7.5 g, 40 mmol), K2C03 (11 g, 80 mmol) in DMF (100 ml) was degassed and stirred at 120 °C under N2 overnight. The reaction mixture was cooled to rt and diluted with EtOAc (200 ml), then washed with saturated brine (200 ml x 3). The organic layer was separated and dried over Na2S04, evaporated to dryness and purified by silica gel
chromatography (petroleum ethers/EtOAc = 10/1) to give the desired product as a white solid (6.2 g, 64 %).
Step (2)
Synthesis of Intermediate 3: A mixture of compound 2 (69.2 g, 1 equiv.), l-chloro-2-iodobenzene (135.7 g, 2 equiv.), Li2C03 (42.04 g, 2 equiv.), K2C03 (39.32 g, 1 equiv.), Cu (1 equiv. 45 μπι) in DMSO (690 ml) was degassed and purged with nitrogen. The resulting mixture was stirred at 140 °C for 36 hours. Work-up of the reaction gave compound 3 at 93 % yield.
Step (3)
Synthesis of Intermediate 4: 2N NaOH (200 ml) was added to a solution of the compound 3 (3.0 g, 9.4 mmol) in EtOH (200 ml). The mixture was stirred at 60 °C for 30min. After evaporation of the solvent, the solution was neutralized with 2N HCl to give a white precipitate. The suspension was extracted with EtOAc (2 x 200 ml), and the organic layer was separated, washed with water (2 x 100 ml), brine (2 x 100 ml), and dried over Na2S04. Removal of solvent gave a brown solid (2.5 g, 92 %).
Step (4)
Synthesis of Intermediate 5: A procedure analogous to the Synthesis of Intermediate 6 in Example 1 was used.
Step (5)
Synthesis of 2-((2-chlorophenyl)(phenyl)amino)-N-(7-(hydroxyamino)-7-oxoheptyl)pyrimidine-5-carboxamide: A procedure analogous to the Synthesis of 2-(diphenylamino)-N-(7-(hydroxyamino)-7-oxoheptyl)pyrimidine-5-carboxamide in Example 1 was used.
Exam le 3: Process development for Steps 2-3 of Example 2

Table 2. Reactants and reagents

(13.8, leq)
(22.2g, 2eq) Cu
5 24.3g (l.Oeq) 47.7g (2.0eq) 240mL 140 °C
K2C03 (1.0 ε¾45μπι)
(19.65, leq)
(42.04g, 2eq) Cu
6 69.2g (l.Oeq) 135.7g (2.0eq) 690mL 140 °C
K2C03 (1.0 ε¾45μπι)
(39.32g, leq)
Table 3. Results

Table 4. Purification of Compound 4 by extraction and slurry

MTBE/Heptane (lOvol/lOvol) 2.83% 2.67% 92.57%
MEK/Heptane (3vol/6vol) 4.42% 3.16% 90.00%
93.48%
EtoAc 3.87% 1.43%
iProAc 3.91% 2.81% 90.91%
Example 4: Improved synthesis of Compound (I)
Reaction Scheme

4 5 (I)
Step (1)
Synthesis of Compound 11: Ethyl 2-chloropyrimidine-5-carboxylate (ACY-5, 7.0 Kgs), ethanol (60 Kgs), 2-Chloroaniline (9.5 Kgs, 2 eq) and acetic acid (3.7 Kgs, 1.6 eq) were charged to a reactor under inert atmosphere. The mixture was heated to reflux. After at least 5 hours the reaction was sampled for HPLC analysis (method TM-113.1016). When analysis indicated reaction completion (< 1% ACY-5), the mixture was cooled to 70 ± 5 °C and N,N-Diisopropylethylamine (DIPEA) was added. The reaction was then cooled to 20 ± 5°C and the mixture was stirred for an additional 2-6 hours. The resulting precipitate is filtered and washed with ethanol (2 x 6 Kgs) and heptane (24 Kgs). The cake is dried under reduced pressure at 50 ± 5 °C to a constant weight to produce 8.4 Kgs compound 11 (81% yield and 99.9% purity (method TM-113.1016)). See 1HNMR data in Figure 3.
Step (2)
Synthesis of Compound 3: Copper powder (0.68 Kgs, 1 eq, <75 micron), potassium carbonate (4.3 Kgs, 3.0 eq), and dimethyl sulfoxide (DMSO, 12.3 Kgs) were added to a reactor (vessel A). The resulting solution was heated to 120 ± 5°C. In a separate reactor (vessel B), a solution of compound 11 (2.9 Kgs) and iodobenzene (4.3 Kgs, 2 eq) in DMSO (5.6 Kgs) was
heated at 40 ± 5°C. The mixture was then transferred to vessel A over 2-3 hours. The reaction mixture was heated at 120 ± 5°C for 8-24 hours, until HPLC analysis (method TM-113.942) determined that < 1% compound 11 was remaining.
Step (3)
Synthesis of Compound 4: The mixture of Step (2) was cooled to 90-100 °C and purified water (59 Kgs) was added. The reaction mixture was stirred at 90-100 °C for 2-8 hours until HPLC (method TM-113.942-see step 2) showed that <1% compound 3 was remaining. The reactor was cooled to 25 °C. The reaction mixture was filtered through Celite, then a 0.2 micron filter, and the filtrate was collected. The filtrate was extracted with methyl t-butyl ether twice (2 x 12.8 Kgs). The aqueous layer was cooled to 0-5 °C, then acidified with 6N hydrochloric acid (HC1) to pH 2-3 while keeping the temperature < 25°C. The reaction was then cooled to 5-15 °C. The precipitate was filtered and washed with cold water. The cake was dried at 45-55 °C under reduced pressure to constant weight to obtain 2.2 kg (65% yield) compound 4 in 90.3% AUC purity (method TM-113.942-see step 2). No dechlorinated product or Cl-migration product (i.e., de-Cl-4 or m-Cl-4) was observed. See 1HNMR data in Figure 4.
Step (4)
Synthesis of Compound 5: Dichloromethane (40.3 Kgs), DMF (33g, 0.04 eq) and compound 4 (2.3 Kg) were charged to a reaction flask. The solution was filtered through a 0.2 μπι filter and was returned to the flask. Oxalyl chloride (0.9 Kgs, 1 eq) was added via addition funnel over 30-120 minutes at < 30 °C. The batch was then stirred at < 30°C until reaction completion (compound 4 <3 %) was confirmed by HPLC (method TM-113.946). Next, the dichloromethane solution was concentrated and residual oxalyl chloride was removed under reduced pressure at < 40 °C. When HPLC analysis (method TM-113.946) indicated that < 0.10%) oxalyl chloride was remaining, the concentrate was dissolved in fresh
dichloromethane (24 Kgs) and transferred back to the reaction vessel (Vessel A).
A second vessel (Vessel B) was charged with Methyl 7-aminoheptanoate
hydrochloride (Compound Al, 1.5 Kgs, 1.09 eq), DIPEA (2.5 Kgs, 2.7 eq), 4
(Dimethylamino)pyridine (DMAP, 42g, 0.05 eq), and DCM (47.6 Kgs). The mixture was cooled to 0-10 °C and the acid chloride solution in Vessel A was transferred to Vessel B while maintaining the temperature at 5 °C to 10 °C. The reaction is stirred at 5-10 °C for 3 to 24 hours at which point HPLC analysis indicated reaction completion (method TM-113.946, compound 4 <5%). The mixture was then extracted with a 1M HC1 solution (20 Kgs), purified water (20 Kgs), 7% sodium bicarbonate (20 Kgs), purified water (20 Kgs), and 25% sodium chloride solution (20 Kgs). The dichloromethane was then vacuumdistilled at < 40 °C and chased repeatedly with isopropyl alcohol. When analysis indicated that <1 mol% DCM was remaining, the mixture was gradually cooled to 0-5 °C and was stirred at 0-5 °C for an at least 2 hours. The resulting precipitate was collected by filtration and washed with cold isopropyl alcohol (6.4 Kgs). The cake was sucked dry on the filter for 4-24 hours, then was further dried at 45-55 °C under reduced pressure to constant weight. 2.2 Kgs (77% yield) was isolated in 95.9% AUC purity (method TM-113.953) and 99.9 wt %. See 1HNMR data in Figure 5.
Step (5)
Synthesis of Compound (I): Hydroxylamine hydrochloride (3.3 Kgs, 10 eq) and methanol (9.6 Kgs) were charged to a reactor. The resulting solution was cooled to 0-5 °C and 25% sodium methoxide (11.2 Kgs, 11 eq) was charged slowly, maintaining the temperature at 0-10 °C. Once the addition was complete, the reaction was mixed at 20 °C for 1-3 hours and filtered, and the filter cake was washed with methanol (2 x 2.1 Kgs). The filtrate (hydroxylamine free base) was returned to the reactor and cooled to 0±5°C. Compound 5 (2.2 Kgs) was added. The reaction was stirred until the reaction was complete (method TM-113.964, compound 5 < 2%). The mixture was filtered and water (28 Kgs) and ethyl acetate (8.9 Kgs) were added to the filtrate. The pH was adjusted to 8 - 9 using 6N HC1 then stirred for up to 3 hours before filtering. The filter cake was washed with cold water (25.7 Kgs), then dried under reduced pressure to constant weight. The crude solid compound (I) was determined to be Form IV/ Pattern D.
The crude solid (1.87 Kgs) was suspended in isopropyl alcohol (IP A, 27.1 Kg). The slurry was heated to 75±5 °C to dissolve the solids. The solution was seeded with crystals of Compund (I) (Form I/Pattern A), and was allowed to cool to ambient temperature. The resulting precipitate was stirred for 1-2 hours before filtering. The filter cake was rinsed with IPA (2 x 9.5 Kgs), then dried at 45-55°C to constant weight under reduced pressure to result in 1.86 kg crystalline white solid Compound (I) (Form I/Pattern A) in 85% yield and 99.5% purity. See 1HNMR data in Figure 6.
Example 5: Alternative synthesis of Compound (I)
Reaction Scheme

(I)
Step (1)
Synthesis of Compound 11: Ethyl 2-chloropyrimidine-5-carboxylate (ACY-5, 250g), ethanol (2179 g), 2-Chloroaniline (339.3 g, 2 eq) and acetic acid (132.1 g, 1.6 eq) were charged to a reactor under inert atmosphere. The mixture was heated to reflux. After at least 5 hours the reaction was sampled for HPLC analysis. When analysis indicated reaction completion (< 1% ACY-5), the mixture was cooled to 70 ± 5 °C and Ν,Ν-Diisopropylethylamine (DIPEA, 553.6 g, 3.2 eq) was added. The reaction was then cooled to 20 ± 5°C and the mixture was stirred for an additional 2-6 hours. The resulting precipitate is filtered and washed with ethanol (2 x 401 g) and heptane (2 x 428 g). The cake is dried under reduced pressure at 50 ± 5 °C to a constant weight to produce 307. lg compound 11 (82.5% yield and 99.7% purity.
Step (2)
Synthesis of Compound 3: Cuprous iodide (17.5g, 8 eq), potassium carbonate (373.8 g, 3 eq), L-Prolin (11.4 g, 0.11 eq.) and dimethyl sulfoxide (DMSO, and 1180 g ) were added to a reactor (vessel A). The resulting solution was heated to 90 ± 5°C. In a separate reactor (vessel B), a solution of compound 11 (250g) and iodobenzene (1469.5 g, 8 eq) in DMSO (402.5 g) was heated at 40 ± 5°C. The mixture was then transferred to vessel A over 2-3 hours. The reaction mixture was heated at 90 ± 5°C for 8-24 hours, until HPLC analysis determined that < 1%) compound 11 was remaining.
Step (3)
Synthesis of Compound 4: The mixture of Step (2) was cooled to 40-50 °C and water (500g) and potassium hydroxide solution 10% (700.0 g, 2.8 eq) were added. The reaction mixture was stirred at 40-50 °C for 2-8 hours until HPLC showed that <1% compound 3 was remaining. The reactor was cooled to 25 °C. The reaction mixture was filtered through Celite, then a 0.2 micron filter, and the filtrate was collected. The filtrate was extracted with toluene (3 x 150g). The aqueous layer was cooled to 0-5 °C, then acidified with hydrochloric acid (HC1) to pH 2-3 while keeping the temperature < 25°C. The reaction was then cooled to 5-15 °C. The precipitate was filtered and washed with cold water. The cake was dried at 45-55 °C under reduced pressure to constant weight to obtain 291 g (81% yield) compound 4 in 98% AUC purity. No dechlorinated product or Cl-migration product (i.e., de-Cl-4 or m-Cl-4) was observed.
Step (4)
Synthesis of Compound 5 :
Compound 4 (250.0 g), A-l (159.2 g, 1.06 eq) and Methy-THF (5113 g) were charged to the reactor. DIPEA (283.7 g, 2.85 eq), hydroxybenzotriazole (HOBt, 12.5 g, 0.11 eq) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC.HC1, 216.3 g, 1.47 eq) were added. The reaction solution was stirred at ambient temperature for 6-24 hours, at which point HPLC analysis indicated reaction completion (compound 4 <3%). The mixture was then extracted with a 1M HC1 solution (2270 g), purified water (2270 g), 7% sodium bicarbonate (2270 g), purified water (2270 g), and 25% sodium chloride solution (2270 g). The Methyl-THF was then vacuumdi stilled at < 40 °C and chased repeatedly with isopropyl alcohol. When analysis indicated that <1 mol% methyl-THF was remaining, the mixture was gradually cooled to 0-5 °C and was stirred at 0-5 °C for an at least 2 hours. The resulting precipitate was collected by filtration and washed with cold isopropyl alcohol (700g). The cake was sucked dry on the filter for 4-24 hours, then was further dried at 45-55 °C under reduced pressure to constant weight. 294g (82% yield) was isolated in 99.6% AUC purity and 99.4 wt %.
Step (5)
Synthesis of Compound (I): Hydroxylamine hydrochloride (330g, 10 eq) and methanol (960g) were charged to a reactor. The resulting solution was cooled to 0-5 °C and 25% sodium methoxide (1120 g, 11 eq) was charged slowly, maintaining the temperature at 0-10 °C. Once
the addition was complete, the reaction was mixed at 20 °C for 1-3 hours and filtered, and the filter cake was washed with methanol (2 x 210 g). The filtrate (hydroxylamine free base) was returned to the reactor and cooled to 0±5°C. Compound 5 (220 g) was added. The reaction was stirred until the reaction was complete (compound 5 < 2%). The mixture was filtered and water (280 g) and ethyl acetate (890 g) were added to the filtrate. The pH was adjusted to 8 -9 using HC1 then stirred for up to 3 hours before filtering. The filter cake was washed with cold water (2570 g), then dried under reduced pressure to constant weight to yield 980 g crude solid in 83% yield. The crude solid compound (I) was determined to be Form IV/ Pattern D.
The crude solid (980 g) was suspended in 1-propanol (400 g) and purified water (220 g). The suspension was heated to 40°C. The batch was then cooled to 38°C over 30 minutes. The solution was seeded with crystals of Compund (I) (Form I/Pattern A, 2-5 wt %). The batch was kept at 37-38°C for 2-4 hours, then was gradually cooled to 20±2°C. Water (950 g) was charged over 3 -5 hours. The batch was cooled to 12°C and was stirred for 2 hrs at this temperature. The batch was filtered and washed with cold 1-propanol/water, then dried at 50±5°C to constant weight to yield 910 g purified compound (I) in 93% yield and 99.8% AUC purity.
“DRUG APPROVALS INT” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent