
Napazimone
https://newdrugapprovals.org/2026/02/17/napazimone/
Anthony Melvin Crasto AfricurePharma Row2Tech Anax AdvectProc Niper-G
Napazimone
https://newdrugapprovals.org/2026/02/17/napazimone/
Anthony Melvin Crasto AfricurePharma Row2Tech Anax AdvectProc Niper-G


BIODEGRADGRADABLE DEVICES, AND METHODS OF MAKING THE SAME
UNITED ARAB EMIRATES UNIVERSITY
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022038578&_gid=202208

جامعة الإمارات العربية المتحدة | |
| Type | Public |
|---|---|
| Established | 1976 |
| Chancellor | Zaki Nusseibeh |
| Vice-Chancellor | Ghaleb Ali Al Hadrami Al Breiki |
| Provost | Mohammed Hassan Ali |
| Undergraduates | 15,019 |
| Postgraduates | 676 |
| Location | , |
| Campus | Urban |
| Nickname | UAEU |
| Website | uaeu |


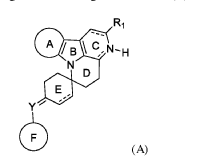
WO2021205354 - NOVEL NATURE-INSPIRED ANTICANCER AND ANTIBACTERIAL MOTIFS AND PHARMACEUTICAL COMPOSITION THEREOF
NOVEL NATURE-INSPIRED ANTICANCER AND ANTIBACTERIAL MOTIFS AND PHARMACEUTICAL COMPOSITION THEREOF
UNIVERSITY OF SHARJAH
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021205354&_cid=P12-KZJPKP-63707-1
WO2021260721 - A NEW COST EFFECTIVE AND SCALABLE PROCESS FOR SYNTHESIZING PURE BRIVARACETAM
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021260721&_cid=P12-KXX1JU-33531-1
EXAMPLES:
EXAMPLE 1: Synthesis of (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7 of scheme A of the present invention]
Example 1 illustrates one pot process for preparing purified (3R)-N-[(1S)-1- carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7] from Intermediate 3 (80% ee) as developed in step 1 of scheme A of the present invention.
Procedure:
In the first step of scheme A of the present invention, a mixture of (R)/(S)-4-propyldihydrofuran-2(3H)-one (Intermediate 3, R: S isomer = 80: 20) (1 eq), (S)-2-aminobutanamide (1.1 eq), triethylamine (1.5 eq) is refluxed at a temperature of 95±5 °C for 24h. The mixture is then cooled to 60-65 °C, washed with a mixture of dichloromethane and diisopropyl ether (2.5 vol) in order to get Intermediate-7 [(3R)-N-[(1S)-1-carbamoyl-propyl]-3-(hydroxymethyl) hexanamide] (80% yield).
Results:
Formation of Intermediate 7 is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 1 depicts: (400 MHz, DMSO-d6): δ 0.6 (t, J= Hz, 6H), 1.07-1.18 (m, 1H), 1.21-1.35 (m, 3H), 1.45-1.43 (m, 1H), 1.61-1.72 (m, 1H), 1.75-1.90 (m, 1H), 2.03 (dd, J=6.64 & 14.08 Hz, 1H), 2.18 (dd, J=7.0 & 14.08 Hz, 1H), 3.28 (t, J=5.36 Hz, 2H), 4.07-4.18 (m, 1H), 4.43 (t, J=5.2 Hz, 1H), 6.95 (s, 1H), 7.28 (s, 1H), 7.76 (d, J=8.08 Hz, 1H).; thus, confirming formation of Intermediate 7 of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 2 provides a (M+H+) value of 231.0; thus, confirming formation of Intermediate 7 of the present invention.
c) The HPLC study is also conducted and the data as graphically illustrated in accompanying figure 3 confirms formation of Intermediate 7 of the present invention with chiral purity of 97.38%
EXAMPLE 2: Preparation of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8A of scheme A of the present invention]: Example 2 illustrates a process for preparing (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8A] from Intermediate 7 of example 1 above as developed in the present invention.
Procedure:
In second step of scheme A of the present invention, the said Intermediate 7 of example 1 above that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(hydroxymethyl) hexanamide (~98% Chemical purity and ~97% Chiral purity) (1736.86 mmol) is dissolved in DCM (1.2 L) at RT into a RBF under N2 atm. Then the solution is cooled to 10-20 C and Oxaloyl chloride (2605.29 mmol) is added to this cooled solution at 10-20 °C. The mixture is stirred for 24 h at 25-40 °C under N2 atm. Completion of the reaction is monitored by TLC. After completion of reaction, the solvent is distilled off and the residual mass is diluted with water (6 L), stirred at 30-50 °C for 4 h. Slurry mass is then filtered and washed with water (2×400 mL) followed by MTBE (800 mL). The solid is dried under vacuum at 50-55 °C for 4-5 h to afford Intermediate 8A that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (92% yield).
Results:
Formation of Intermediate 8A is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 4 depicts: 1H NMR (400 MHz, DMSO-d6) : δ 0.83 (t, J=7.44 Hz, 3H), 0.85 (t, J=6.72 Hz, 3H), 1.20-1.40 (m, 4H), 1.43-1.56 (m, 1H), 2.08-2.18 (m, 1H), 2.20-2.28 (m, 2H), 3.61 (dd, J=4.6 & 10.8 Hz, 1 H), 3.67 (dd, J=4.6 & 10.8 Hz, 1H), 4.07-4.18 (m, 1H), 6.95 (s, 1H), 7.29 (s, 1H), 7.89 (d, J=8.12 Hz, 1H); thus confirming formation of Intermediate 8A of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 5 (a, b) provides a (M+H+) value of 249.20; thus, confirming formation of Intermediate 8A of the present invention.
EXAMPLE 3: Process for purification of Intermediate 8A forming Intermediate 8B Example 3 illustrates a process for purifying the said Intermediate 8A of example 2 above of the present invention.
Procedure:
The Intermediate 8A as obtained in example 2 above [that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide] is first dissolved in a polar solvent like Acetonitrile raising the temperature to 50 to 60 °C; followed by stirring and then addition of another solvent methyl tert-butyl ether (MTBE) which is less polar in nature. The mixture is then cooled down to 0°C, the filtered mass thus obtained is dried in order to obtain a white solid of Intermediate 8B. The material thus obtained is further dissolved in THF (5 vol) at 60 °C, cooled to 20-30°C, followed by addition of heptane (5 vol), stirred at 10°C to 30 °C for 1 h. The mass obtained is filtered and washed with heptane (2×1 vol), dried under vacuum at 50-55°C in order to afford formation of purer form of Intermediate 8A that is Intermediate 8B that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid having a chemical purity of 99.9% along with a chiral purity of 100% (yield: 390 g).
Results:
The purification of Intermediate 8A is further confirmed by the following analytical test results:
a) Chiral HPLC: A Chiral HPLC as illustrated in accompanying figure 6 confirmed formation of purest form of Intermediate 8B having 100% chiral purity [Peak 1; RT (min) = 6.244; %Area=100%].
b) GLP-HPLC: A GLP-HPLC as illustrated in accompanying figure 7 further confirmed formation of Intermediate 8B having 99.9% chemical purity [Peak 3; BRIV8; RT = 29.278; % Area=99.90%].
EXAMPLE 4: Synthesis of (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7’ of scheme B of the present invention]
Example 4 illustrates one pot process for preparing purified (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7’] from Intermediate 6 (99.99% ee) as developed in step-1 scheme B of the present invention.
Procedure:
In another method, in the first step of scheme B of the present invention, a mixture of (R)/(S)-4-propyldihydrofuran-2(3H)-one (Intermediate 6: S isomer = 99.99% : 0.1%) (1 eq), (S)-2-aminobutanamide (1.7 eq), triethylamine (5 eq) is refluxed at a temperature between 95±5 °C for 24 h. Then, the crude reaction mass is cooled and washed with dichloromethane and diisopropyl ether mixture (2.5 vol) in order to achieve Intermediate-7’ of scheme B of the present invention [(3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide] (90% yield).
Results:
The chiral purity of the formed Intermediate 7’ is analyzed by HPLC method and the data as graphically illustrated in accompanying figure 8 confirms formation of Intermediate 7’ of the present invention with chiral purity of 99.11%
EXAMPLE 5: Preparation of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8’ of scheme B of present invention]: Example 5 illustrates a process for preparing purest form of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8’] from Intermediate 7’ of example 4 as developed in scheme B (step 2) of the present invention.
Procedure:
In the second step of scheme B of the present invention, the intermediate 7’ of the above example 4 that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(hydroxymethyl) hexanamide (98% Chemical purity and >99%Chiral purity) (1736.86 mmol) is dissolved in DCM (1.2 L) at RT in a round bottomed flask under N2 atm. Then the solution is cooled to 10-30 °C and 1-Chloro-N,N,2-trimethyl-1-propenylamine (2605.29 mmol) is added to this cooled solution at 10-30 °C. The mixture is stirred for 24 h at 25-40 °C under N2 atm. Completion of the reaction is monitored by TLC. After completion of the reaction, the solvent is distilled off and the residual mass is diluted with water (6 L), stirring at 30-50 °C for 4 h. The slurry mass thus obtained is then filtered and washed with water (2×400 mL) followed by methyl tert-buty ether (MTBE) (800 mL). The solid thus obtained is dried under vacuum at 50-55 °C for 4-5 h in order to afford formation of Intermediate 8’ that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (92% yield).
Results:
Formation of Intermediate 8’ is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 9 depicts: (400 MHz, DMSO-d6): 1H NMR (400 MHz, DMSO-d6) : δ 0.83 (t, J=7.44 Hz, 3H), 0.85 (t, J=6.72 Hz, 3H), 1.20-1.40 (m, 4H), 1.43-1.56 (m, 1H), 2.08-2.18 (m, 1H), 2.20-2.28 (m, 2H), 3.61 (dd, J=4.6 & 10.8 Hz, 1 H), 3.67 (dd, J=4.6 & 10.8 Hz, 1H), 4.07-4.18 (m, 1H), 6.95 (s, 1H), 7.29 (s, 1H), 7.89 (d, J=8.12 Hz, 1H); thus, confirming formation of Intermediate 8’ of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 10 provides a (M+H+) value of 249.1; thus, confirming formation of Intermediate 8’ of the present invention.
c) The HPLC data as illustrated in accompanying figure 11 confirms 100% chiral purity of Intermediate 8’.
EXAMPLE 6: Preparation of (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide [Brivaracetam-API]:
Example 6 illustrates a process for preparing (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide [Brivaracetam API] from Intermediate 8B of example 3 or from Intermediate 8’ of example 5 as developed in step 3 of scheme A or scheme B of the present invention respectively.
Procedure:
In the final step of scheme A or scheme B of the present invention, the intermediate 8B of example 3 or intermediate 8’ of example 5 above that is (3R)-N-((1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide (1608.04 mmol) is dissolved in dimethyl acetamide (0.5 vol) and isopropylacetate (2 L) into a RBF at 25-30 °C under N2 atm. Then 18-Crown-6 (160.79 mmol) is added into the solution and stirred at RT for 30 min. Reaction mixture is then cooled to 0-10 °C and t-BuOK (1.5 eq) is added portion wise to the cooled solution over 1 h maintaining the temperature from - 0-10 °C to 25 °C under N2 atm. Stirring is then continued for 2 h at -10 °C to 0 °C and then for 12 h at 15-25 °C under N2 atm. Completion of reaction is monitored by TLC. After completion of reaction, the reaction mixture is quenched with addition of 1M HCl solution (pH~6.5-7.0). The resulting mixture is extracted with i-PrOAc (2 L) and MTBE (1 L). Water (0.5 L) is added to the combined organic extract and then filtered through celite bed, washed the bed with MTBE-i-PrOAc (1:1) (400 mL). The organic part is separated and the aqueous part is re-extracted with i-PrOAc-MTBE (1:1) (2 ×0.8 L). The combined organic phases are washed with brine solution (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuum under a rotary evaporator to afford crude API. Distillation of dimethylacetamide solvent from the crude is then done at high vacuum pressure (0.05 mm Hg) at 70 °C. Crude product is then dissolved in isopropyl acetate (1.6 L) and treated with activated charcoal (7% w/w) to afford a tech-grade crude of Brivaracetam API as a white solid (yield: 90%) with 97.82% chemical purity.
Results:
Formation of Brivaracetam API is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 12 depicts: 1H NMR (400 MHz, DMSO-d6) : δ 0.77 (t, J=7.32 Hz, 3H), 0.87 (t, J=7.2 Hz, 3H), 1.21-1.31 (m, 2H), 1.33-1.43 (m, 2H), 1.50-1.62 (m, 1H), 1.73-1.84 (m, 1H), 1.97 (dd, J=8.0 & 16.12 Hz, 1H), 2.18-2.28 (m, 1H), 2.37 (dd, J=8.4 & 16.14 Hz, 1H), 3.11 (dd, J=7.16 & 9.44 Hz, 1H), 3.36 (dd, J=9.2 & 17.5 Hz, 1H), 4.30 dd, J=5.44 & 10.28 Hz, 1H), 6.98 (s, 1H), 7.32 (s, 1H); thus, confirming formation of Brivaracetam API of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 13 provides a (M+H+) value of 213.0; thus, confirming formation of Brivaracetam API of the present invention.
^ Purification of Brivaracetam API:
The Brivaracetam thus formed above is further purified by means of dissolving the said material (307 g) in 30% i-PrOAc -MTBE (1 vol) at 55-60 °C, cool to 20-30°C. A mixture of Heptane and MTBE and DIPE (2:2:1) is added, stirred at 10 °C to 30°C for 1 h. The obtained mass is filtered and washed with heptane, which is subsequently dried under vacuum at 40-45 °C to afford (3R)-N-((1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (yield: 80%, chiral purity 99.93% and chemical purity 99.94%).
Results:
a) Chiral HPLC: A Chiral HPLC as illustrated in accompanying figure 14 confirmed formation of purest form of Brivaracetam API having 99.93% chiral purity [Peak 2; RT (min) = 9.45; %Area=99.93%].
b) GLP-HPLC: A GLP-HPLC as illustrated in accompanying figure 15 further confirmed formation of Brivaracetam API having 99.9% chemical purity [Peak 2; RT = 21.138; % Area=99.94%].
NEW PATENTS
1 WO/2021/258625ANTI-SAR-COV-2 (COVID-19) FULLY HUMANIZED MONOCLONAL ANTIBODY, AND PREPARATION METHOD THEREFOR AND APPLICATION THEREOF
SHENZHEN INSTITUTES OF ADVANCED TECHNOLOGY CHINESE ACADEMY OF SCIENCES
2 WO/2021/260164THROMBIN C-TERMINAL PEPTIDES TO TREAT CORONAVIRAL INFECTIONS
IN2CURE AB
3 WO/2021/211332METHODS AND KITS FOR DETECTING OR DETERMINING AN AMOUNT OF AN ANTI-Β-CORONAVIRUS ANTIBODY IN A SAMPLE
ABBOTT LABORATORIES
4 WO/2021/262744KITS AND METHODS FOR ISOTHERMALLY AMPLIFYING RIBONUCLEIC ACID OF SEVERE ACUTE RESPIRATORY VIRUS COV2
UNIVERSITY OF WASHINGTON
5 WO/2021/262894METHODS FOR DIAGNOSING RESPIRATORY PATHOGENS AND PREDICTING COVID-19 RELATED OUTCOMES
THE REGENTS OF THE UNIVERSITY OF COLORADO, A BODY CORPORATE
////////////////
(S,Z)-3,7-DIMETHYLNON-6-EN-1-OL AND ITS USE AS FRAGRANCE
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021198525&_gid=202140
GIVAUDAN SA
USES OF CYCLOCARYA PALIURUS LEAF FLAVONE EXTRACT IN PREPARATION OF ANTIBACTERIAL MEDICAMENTS AND/OR ANTIBACTERIAL AGENTS
WEST CHINA HOSPITAL, SICHUAN UNIVERSITY
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021196505&_gid=202140
NEW PATENT
WO/2021/180078 METHOD FOR PREPARING CHITOSAN USING SNOW CRAB SHELLS


