
US 7902380, Levetiracetam
http://www.google.im/patents/US7902380
preparation of both the (S)— and (R)-enantiomers of alpha-ethyl-2-oxo-1-pyrrolidineacetamide of formula 1 from (RS)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid of formula 2.

The following is an exemplary scheme of the process:

Suitable resolving agents include optically pure bases such as alpha-methylbenzylamine and dehydroabietylamine, of which alpha-methylbenzylamine is preferred. (S)-2 can be prepared by forming the salt with (R)-alpha-methylbenzylamine and the (R)-2 can be prepared by forming the salt with (S)-alpha-methylbenzylamine.
NOTE......R)-alpha-methylbenzylamine is desired agent to get levetiracetam
The optical resolution of 2 may be carried out by, for example, the formation of a salt of (S)-2 with the optically active base (R)-alpha-methylbenzylamine or dehydroabietylamine (S. H. Wilen et al. Tetrahedron, 33, (1997), 2725-2736). Likewise, the (R)-2 can be prepared by forming the salt with (S)-alpha-methylbenzylamine. The racemic (RS)-2 used as starting material can be prepared by the known procedure described in GB 1309692.
Surprisingly we have found that the undesired (R) or (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid or their mixture can be epimerized by treating it with an acid anhydride, preferably acetic anhydride, propionic anhydride and butyric anhydride, to furnish a mixture of (R) and (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid in excellent yield. The recovered (RS)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid can be optically resolved by the same procedure above. In this way, we are able to obtain almost complete conversion of the (RS)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid to the desired (R) or (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid.


The process is depicted below:

A suspension of (s)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid (45 g, 0.26 mol) in methylene chloride (225 ml) was cooled to 0° C. and triethylamine (53 g, 0.53 mol) and methanesulfonyl chloride (39 g, 0.34 mol) were added dropwise. The mixture was stirred at 0° C. for 30 min., then a stream of ammonia was purged in the solution for 2 hours. The insoluble solids were filtered and the filtrate was concentrated. The product was crystallized from methyl isobutyl ketone to give 36 g (80%) of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide.
A suspension of (R)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid (35 g, 0.20 mol) in methylene chloride (225 ml) was cooled to 0° C. and triethylamine (41 g, 0.40 mol) and methanesulfonyl chloride (29 g, 0.26 mol) were added dropwise. The mixture was stirred at 0° C. for 30 min., then a stream of ammonia was purged in the solution at 0° C. for 2 hours. The insoluble solids were filtered and the filtrate was concentrated. The product was recrystallized from methyl isobutyl ketone to give 27.5 g (78%) of (R)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide.
A solution of (R)-alpha-methylbenzylamine (106 g) and triethylamine (89 g) in toluene (100 ml) was added to a suspension of (RS)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid (300 g, 1.75 mol) in toluene (1 L). The mixture was heated until complete dissolution, cooled to room temperature and stirred for 3 hours. The solids were filtered and rinsed with toluene (300 ml) to give 250 g of (s)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid (R)-alpha-methylbenzylamine salt. The solids were crystallized from toluene and 205 g (yield 41%) of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid (R)-alpha-methylbenzylamine salt was obtained. The isolated solid was treated with hydrochloric acid solution and the enantiomerically pure (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid could be isolated in 90% yield.
The combined mother liquors from above were concentrated to half volume and water (200 ml) and 50% sodium hydroxide (52 g) were added sequentially and the mixture was stirred at 20° C. for 30 min. and then was separated. The aqueous layer was washed with toluene (150 ml), acidified with 32% hydrochloric acid until pH=2-3. The resulting suspension was cooled to 0-5° C. and stirred for 2 h. The solids were collected by filtration, and were rinsed with cold water. The damp solids were dried under vacuum oven at 40-50° C. for 4 h to give 160 g of (R)-enriched ethyl-2-oxo-1-pyrrolidineacetic acid. To the above solids, toluene (640 ml) and acetic anhydride (145 g) were added and the mixture was heated to reflux for 10 h. The solution was cooled to 20° C. and stirred for another 2 h. The solids were collected by filtration and rinsed with toluene (150 ml) to give (RS)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid (152 g).
extras

(R)-(+)-alpha-methyl-benzylamine
Levetiracetam industrial process

2 pyrolidinone
ethyl 2 bromo butyrate
(R)-(+)-alpha-methyl-benzylamine
ethyl chloro formate
US4943639.
cut paste
note………….racemic (±)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid is obt by rxn of 2 pyrolidinone with ethyl 2 bromo acetate

+/-)-(R,S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid methyl ester. CAS# 33978-83-5

EXAMPLE 1 (a) Preparation of the (R)-alpha-methyl-benzylamine salt of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid
8.7 kg (50.8 moles) of racemic (±)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid are suspended in 21.5 liters of anhydrous benzene in a 50 liter reactor. To this suspension is added gradually a solution containing 3.08 kg (25.45 moles) of (R)-(+)-alpha-methyl-benzylamine and 2.575 kg (25.49 moles) of triethylamine in 2.4 liters of anhydrous benzene. This mixture is then heated to reflux temperature until complete dissolution It is then cooled and allowed to crystallize for a few hours. 5.73 kg of the (R)-alpha-methyl-benzylamine salt of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid are thus obtained.
Melting point: 148°-151° C. Yield: 77.1%.
This salt may be purified by heating under reflux in 48.3 liters of benzene for 4 hours. The mixture is cooled and filtered to obtain 5.040 kg of the desired salt. Melting point: 152°-153.5° C. Yield: 67.85%.
(b) Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid
5.04 kg of the salt obtained in (a) above are dissolved in 9 liters of water. 710 g of a 30% sodium hydroxide solution are added slowly so that the pH of the solution reaches 12.6 and the temperature does not exceed 25° C. The solution is stirred for a further 20 minutes and the alpha-methylbenzylamine liberated is extracted repeatedly with a total volume of 18 liters of benzene.
The aqueous phase is then acidified to a pH of 1.1 by adding 3.2 liters of 6N hydrochloric acid. The precipitate formed is filtered off, washed with water and dried.
The filtrate is extracted repeatedly with a total volume of 50 liters of dichloromethane. The organic phase is dried over sodium sulfate and filtered and evaporated to dryness under reduced pressure.
The residue obtained after the evaporation and the precipitate isolate previously, are dissolved together in 14 liters of hot dichloromethane. The dichloromethane is distilled and replaced at the distillation rate, by 14 liters of toluene from which the product crystallizes.
The mixture is cooled to ambient temperature and the crystals are filtered off to obtain 2.78 kg of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid.
Melting point: 125.9° C. [alpha]D 20 =-26.4° (c=1, acetone). Yield: 94.5%.
(c) Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide
34.2 g (0.2 mole) of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid are suspended in 225 ml of dichloromethane cooled to -30° C. 24.3 g (0.24 mole) of triethylamine are added dropwise over 15 minutes. The reaction mixture is then cooled to -40° C. and 24.3 g (0.224 mole) of ethyl chloroformate are added over 12 minutes. Thereafter, a stream of ammonia is passed through the mixture for 41/2 hours. The reaction mixture is then allowed to return to ambient temperature and the ammonium salts formed are removed by filtration and washed with dichloromethane. The solvent is distilled off under reduced pressure. The solid residue thus obtained is dispersed in 55 ml toluene and the dispersion is stirred for 30 minutes and then filtered. The product is recrystallized from 280 ml of ethyl acetate in the presence of 9 g of 0,4 nm molecular sieve in powder form.
24.6 g of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide are obtained.
Melting point: 115°-118° C. [alpha]D 25 =-89.7° (c=1, acetone). Yield: 72.3%.
Analysis for C8 H14 N2 O2 in % calculated: C 56.45. H 8.29. N 16.46. found: 56.71. 8.22. 16.48.
The racemic (±)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid used in this synthesis has been prepared in the manner described below.
A solution containing 788 g (19.7 moles) of sodium hydroxide in 4.35 liters of water is introduced over 2 hours into a 20 liter flask containing 3.65 kg (18.34 moles) of ethyl (±)-alpha-ethyl-2-oxo-1-pyrrolidineacetate at a temperature not exceeding 60° C. When this addition is complete, the temperature of the mixture is raised to 80° C. and the alcohol formed is distilled off until the temperature of the reaction mixture reaches 100° C.
The reaction mixture is then cooled to 0° C. and 1.66 liter (19.8 moles) of 12N hydrochloric acid is added over two and a half hours. The precipitate formed is filtered off, washed with 2 liters of toluene and recrystallized from isopropyl alcohol. 2.447 kg of racemic (±)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid, melting at 155°-156° C., are thus obtained. Yield: 78%.
Analysis for C8 H13 NO3, in % calculated: C 56.12. H 7.65. N 8.18. found: 55.82. 8.10. 7.97.
EXAMPLE 2 (a) Preparation of ethyl (S)-4-[[1-(aminocarbonyl)propyl]amino]butyrate
143.6 ml (1.035 mole) of triethylamine are added to a suspension of 47.75 g (0.345 mole) of (S)-2-amino-butanamide hydrochloride ([alpha]D 25 : +26.1°; c=1, methanol) in 400 ml of toluene. The mixture is heated to 80° and 67.2 g (0.345 mole) of ethyl 4-bromobutyrate are introduced dropwise.
The reaction mixture is maintained at 80° C. for 10 hours and then filtered hot to remove the triethylamine salts. The filtrate is then evaporated under reduced pressure and 59 g of an oily residue consisting essentially of the monoalkylation product but containing also a small amount of dialkylated derivative are obtained.
The product obtained in the crude state has been used as such, without additional purification, in the preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide by cyclization.
(b) Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide
54 g of the crude product obtained in a) above are dissolved in 125 ml of toluene in the presence of 2 g of 2-hydroxypyridine. The mixture is heated at 110° C. for 12 hours.
The insoluble matter is filtered off hot and the filtrate is then evaporated under reduced pressure.
The residue is purified by chromatography on a column of 1.1 kg of silica (column diameter: 5 cm; eluent: a mixture of ethyl acetate, methanok and concentrated ammonia solution in a proportion by volume of 85:12:3).
The product isolated is recrystallized from 50 ml of ethyl acetate to obtain 17.5 g of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide.
Melting point: 117° C. [alpha]D 25 : -90.0° (c=1, acetone). Yield: 41%.
EXAMPLE 3 (a) Preparation of (S)-N-[1(aminocarbonyl)propyl]-4-chlorobutanamide
345.6 g (2.5 moles) of ground potassium carbonate are mixed with 138.5 g (1 mole) of (S)-2-amino-butanamide hydrochloride in 2.5 liters of acetonitrile. The reaction mixture is cooled to 0° C. and a solution of 129.2 g (1.2 mole) of 4-chlorobutyryl chloride in 500 ml of acetonitrile is introduced dropwise. After the addition, the reaction mixture is allowed to return to ambient temperature; the insoluble matter is filtered off and the filtrate evaporated under reduced pressure. The crude residue obtained is stirred in 1.2 liter of anhydrous ether for 30 minutes at a temperature between 5° and 10° C. The precipitate is filtered off, washed twice with 225 ml of ether and dried in vacuo to obtain 162.7 g of (S)-N-[1-(aminocarbonyl)propy]-4-chlorobutanamide.
Melting point: 118°-123° C. [alpha]D 25 : -18° (c=1, methanol). Yield: 78.7%.
The crude product thus obtained is very suitable for the cyclization stage which follows. It can however be purified by stirring for one hour in anhydrous ethyl acetate.
Melting point: 120°-122° C. [alpha]D 25 : -22.2° (c=1, methanol).
(b) Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide
6.2 g (0.03 mole) of (S)-N-[1(aminocarbonyl)propyl]-4-chlorobutamine and 0.484 g (0.0015 mole) of tetrabutylammonium bromide are mixed in 45 ml of dichloromethane at 0° C. under a nitrogen atmosphere. 2.02 g (0.036 mole) of potassium hydroxide powder are added over 30 minutes, at such a rate that the temperature of the reaction mixture does not exceed +2° C. The mixture is then stirred for one hour, after which a further 0.1 g (0.0018 mole) of ground potassium hydroxide is added and stirring continued for 30 minutes at 0° C. The mixture is allowed to return to ambient temperature. The insoluble matter is filtered off and the filtrate is concentrated under reduced pressure. The residue obtained is recrystallized from 40 ml of ethyl acetate in the presence of 1.9 g of 0,4 nm molecular sieve. The latter is removed by hot filtration to give 3.10 g of (S)-alphaethyl-2-oxo-1-pyrrolidineacetamide.
Melting point: 116.7° C. [alpha]D 25 : -90.1° (c=1, acetone). Yield: 60.7%.
EXAMPLE 4 Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide……levetiracetam
This example illustrates a variant of the process of Example 3, in which the intermediate 4-chlorobutanamide obtained in situ is not isolated. 84 g of anhydrous sodium sulfate are added to a suspension of 69.25 g (0.5 mole) of (S)-2-amino-butanamide hydrochloride in 600 ml of dichloromethane at ambient temperature. The mixture is cooled to 0° C. and 115 g of ground potassium hydroxide are added, followed by 8.1 g (0.025 mole) of tetrabutylammonium bromide dissolved in 100 ml of dichloromethane. A solution of 77.5 g of 4-chlorobutyryl chloride in 100 ml of dichlorometha is added dropwise at 0° C., wih vigorous stirring. After 5 hours’ reaction, a further 29 g of ground potassium hydroxide are added. Two hours later, the reaction mixture is filtered over Hyflo-cel and the filtrate evaporated under reduced pressure. The residue (93.5 g) is dispersed in 130 ml of hot toluene for 45 minutes. The resultant mixture is filtered and the filtrate evaporated under reduced pressure. The residue (71.3 g) is dissolved hot in 380 ml of ethyl acetate to which 23 g of 0,4 nm molecular sieve in powder form are added. This mixture is heated to reflux temperature and filtered hot. After cooling the filtrate, the desired product crystallizes to give 63 g of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide.
Melting point: 117° C. [alpha]D 25 : -91.3° (c=1, acetone). Yield: 74.1%.
—
FROM MY OLD POST
(±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide a key levetiracetam intermediate

(±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
methyl (±)-(R,S)-alpha-ethyl-2-oxo-l -pyrrolidine acetate with (+)-(R)-(l-phenylethyl)- amine in toluene in the presence of a base such as sodium hydride or methoxide; crystallization- induced dynamic resolution of the resultant (±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide

(R)-(+)-1-Phenylethylamine
33978-83-5
1-Pyrrolidineacetic acid, α-ethyl-2-oxo-, methyl ester
1-Pyrrolidineacetic acid, α-ethyl-2-oxo-, methyl ester

1004767-60-5
1-Pyrrolidineacetamide, α-ethyl-2-oxo-N-[(1R)-1-phenylethyl]-
(±)-(R.S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
1-Pyrrolidineacetamide, α-ethyl-2-oxo-N-[(1R)-1-phenylethyl]-
(±)-(R.S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
Example 1
(±)-(R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide.
In a 100 ml reactor equipped with mechanical stirring, thermometer and bubble condenser, 13.4 g of (±)-(R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester (71.6 mmol), 8.8 g of (+)-(R)-(l-phenylethyl)-amine (72.5 mmol) and 45 ml of tetrahydrofuran were charged. 3.4 g of NaH (60% dispersion in mineral oil, 85.6 mmol) was added in small portions under nitrogen atmosphere. Reaction mixture was maintained at room temperature for about 2 h. Then, it was heated up to 350C and kept under stirring overnight. Reaction was controlled by TLC (Rf = 0.5, AcOEt/silica gel).
At reaction completed, one night at 35°C temperature, reaction mixture was cooled to room temperature and 30 ml of water was slowly charged. It was transferred into a separatory funnel and was diluted with 30 ml of water and 80 ml of dichloromethane. Phases were separated and the aqueous one was washed with 50 ml of dichloromethane. Collected organic phases were washed with an aqueous acid solution, dried on Na2SO4, filtered and concentrated under vacuum. 19.5 g of an oil residue was obtained which slowly solidified. Solid was suspended in 20 ml of a hexane/dichloromethane 9/1 v/v mixture. It was then filtered, washed with 10 ml of the same solvent mixture and dried at 400C to give 12.1 g of the title compound (44.1 mmol, 61.6% yield) as dry solid.
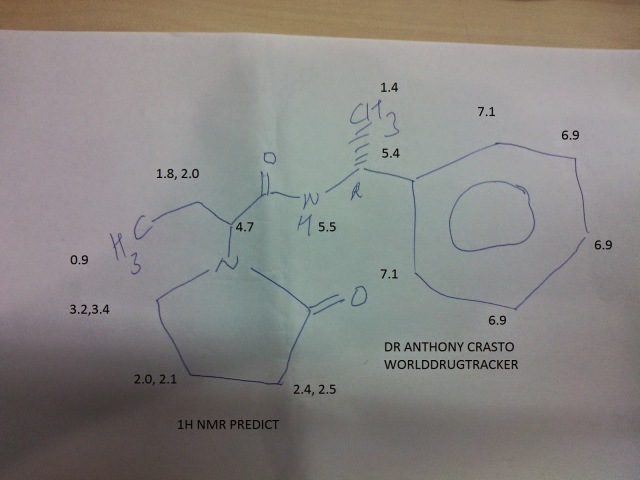
1H NMR (400.13 MHz, CDCl3, 25 0C): δ (ppm, TMS)
7.35-7.19 (1OH, m),
6.49 (2H, br s),
5.09-5.00 (2H, m),
4.41 (IH, dd, J = 8.3, 7.4 Hz),
4.36 (IH, dd, J = 8.6, 7.1 Hz),
3.49 (IH, ddd, J = 9.8, 7.7, 6.6 Hz),
3.41 (IH, ddd, J = 9.8, 7.7, 6.2 Hz),
3.30 (IH, ddd, J = 9.6, 8.3, 5.5 Hz),
3.13 (IH, ddd, 9.7, 8.5, 6.1 Hz), 2.47-2.38 (2H, m), 2.41 (IH, ddd, J = 17.0, 9.6, 6.3 Hz), 2.26 (IH, ddd, 17.0, 9.5, 6.6 Hz), 2.10-1.98 (2H, m), 2.01-1.89 (IH, m), 1.99-1.88 (IH, m), 1.98-1.85 (IH, m), 1.88-1.78 (IH, m), 1.75- 1.62 (IH, m), 1.72-1.59 (IH, m), 1.45 (3H, d, J = 7.1 Hz), 1.44 (3H, d, J = 7.1 Hz), 0.90 (3H, t, J = 7.4 Hz), 0.86 (3H, t, J = 7.4 Hz).


7.35-7.19 (1OH, m),
6.49 (2H, br s),
5.09-5.00 (2H, m),
4.41 (IH, dd, J = 8.3, 7.4 Hz),
4.36 (IH, dd, J = 8.6, 7.1 Hz),
3.49 (IH, ddd, J = 9.8, 7.7, 6.6 Hz),
3.41 (IH, ddd, J = 9.8, 7.7, 6.2 Hz),
3.30 (IH, ddd, J = 9.6, 8.3, 5.5 Hz),
3.13 (IH, ddd, 9.7, 8.5, 6.1 Hz), 2.47-2.38 (2H, m), 2.41 (IH, ddd, J = 17.0, 9.6, 6.3 Hz), 2.26 (IH, ddd, 17.0, 9.5, 6.6 Hz), 2.10-1.98 (2H, m), 2.01-1.89 (IH, m), 1.99-1.88 (IH, m), 1.98-1.85 (IH, m), 1.88-1.78 (IH, m), 1.75- 1.62 (IH, m), 1.72-1.59 (IH, m), 1.45 (3H, d, J = 7.1 Hz), 1.44 (3H, d, J = 7.1 Hz), 0.90 (3H, t, J = 7.4 Hz), 0.86 (3H, t, J = 7.4 Hz).


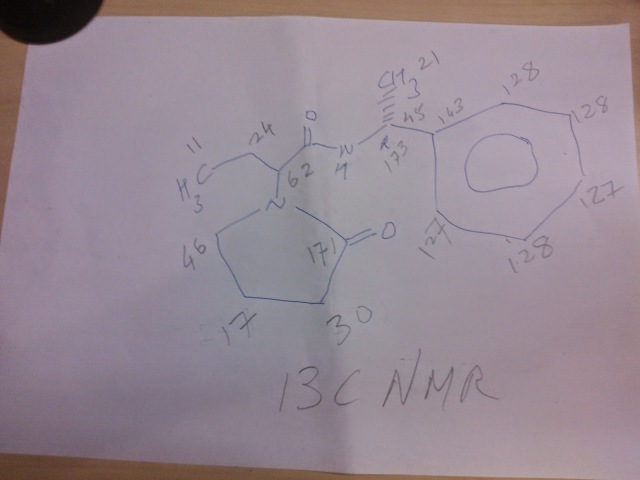
13C NMR (100.62 MHz, CDCl3, 25 0C): δ (ppm, TMS)
176.05 (CO), 176.00 (CO), 169.08 (CO),
168.81 (CO), 143.59 (Cquat),
143.02 (Cquat), 128.66 (2 x CH), 128.55 (2 x CH),
127.33 (CH), 127.19 (CH), 126.05 (2 x CH),
125.80 (2 x CH), 56.98 (CH), 56.61 (CH),
48.90 (CH), 48.84 (CH), 44.08 (CH2),
43.71 (CH2), 31.19 (CH2), 31.07 (CH2), 22.08 (CH3),
22.04 (CH3), 21.21 (CH2), 20.68 (CH2),
18.28 (CH2), 18.08 (CH2), 10.50 (CH3), 10.45 (CH3).
176.05 (CO), 176.00 (CO), 169.08 (CO),
168.81 (CO), 143.59 (Cquat),
143.02 (Cquat), 128.66 (2 x CH), 128.55 (2 x CH),
127.33 (CH), 127.19 (CH), 126.05 (2 x CH),
125.80 (2 x CH), 56.98 (CH), 56.61 (CH),
48.90 (CH), 48.84 (CH), 44.08 (CH2),
43.71 (CH2), 31.19 (CH2), 31.07 (CH2), 22.08 (CH3),
22.04 (CH3), 21.21 (CH2), 20.68 (CH2),
18.28 (CH2), 18.08 (CH2), 10.50 (CH3), 10.45 (CH3).


Example 2 (±)-(R.S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide (alternative 1).
In a 500 ml reactor equipped with mechanical stirring, thermometer and condenser, 24.2 g of (+)-(R)-(l-phenylethyl)-amine (199.51 mmol) and 40 ml of toluene were charged. By keeping the reaction mixture at 00C temperature under nitrogen atmosphere, 9.5 g of NaH (60% mineral oil suspension, 237.50 mmol) was added in small portions. At the same temperature, 190.0 g of a toluene solution of (±)-(R,S)- alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester (19.28% equal to 36.63 g, 197.77 mmol) was charged. Reaction mixture was then heated up to 35°C and maintained in that condition till complete disappearing of methyl ester reagent (about 14 h; checked by HPLC).
At reaction completed, reaction mixture was cooled and when room temperature was reached, 100 ml of water was slowly charged. Aqueous phases were separated and extracted with toluene (2 x 75 ml). Collected organic phases were treated with acid water till neuter pH. Solvent was evaporated and residue was suspended in about 100 ml of heptane for about 30 minutes. Product was isolated by filtration and dried in oven at 400C temperature under vacuum overnight to give 45.2 g of the title compound (164.54 mmol, 83.2% yield, d.e. 0.0%) as white dusty solid.
Example 3
(±)-(R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide (alternative 2).
In a 500 ml reactor equipped with mechanical stirring, thermometer and Dean-Stark distiller, 24.2 g of (+)-(R)-(l-phenylethyl)-amine (199.51 mmol) and 40 ml of toluene were charged. By keeping the reaction mixture at 00C temperature, 42.7 g of sodium methoxide (30% solution in methanol, 237.14 mmol) was added under nitrogen atmosphere. At the same temperature, 190.0 g of a toluene solution of (±)- (R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester (19.28% equal to 36.63 g, 197.77 mmol) was charged. Reaction mixture was then heated up to 65- 700C and maintained in that condition till complete disappearing of methyl ester reagent (about 4 h; checked by HPLC). After a work-up carried out according to the procedure described in example 2, 40.2 g of the title compound (146.53 mmol, 74.1% yield, d.e. 0.0%) as white dusty solid was obtained.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE











































