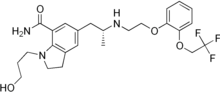Acitretin
PDT PATENT US4105681
Process for preparation of acitretin
Emcure Pharmaceuticals Ltd
EMCURE PHARMACEUTICALS LIMITED [IN/IN]; an Indian company at EMCURE HOUSE, T-184, MIDC., Bhosari, Pune - 411 026 Maharashtra (IN)
GURJAR MUKUND KESHAV; (IN).
JOSHI SHASHIKANT GANGARAM; (IN).
BADHE SACHIN ARVIND; (IN).
KAMBLE MANGESH GORAKHANATH; (IN).
MEHTA SAMIT SATISH; (IN)
The present invention Provides a process for preparation of {(2E, 4E, 6E, 8E) -9- (4-methoxy-2,3,6-trimethyl) phenyl-3,7-dimethyl-nona-2,4,6 , 8} tetraenoate, acitretin year intermediate of formula (VI) with trans isomer ≥97%, comprenant of Reacting 3-formyl-Crotonic acid butyl ester of formula (V) Substantially free of impurities, with 5- (4-methoxy- 2,3,6-trimethylphenyl) -3-methyl-penta-2,4-diene-l-triphenyl phosphonium bromide of formula (IV) and isolating resulting compound of formula (VI) Treating the filtrate with iodine for isomerization of the Undesired cis intermediate and finally Obtaining acitretin (I), with trans isomer Desired ≥97%.
Samit Satish Mehta holds the position of the President - Research & Development
Acitretin of formula (I), chemically known as (2E,4E,6E,8E)-9-(4-methoxy-2,3,6- trimethyl)phenyl-3,7-dimethyl-nona-2,4,6,8-tetraenoic acid, is a second generation retinoid a roved by USFDA in 1996, for the treatment of psoriasis.
Acitretin (I)
The process for preparation of acitretin (I) was first disclosed in US 4,105,681 wherein the intermediate, 5-(4-methoxy-2,3,6-trimethylphenyl)-3-methyl-penta-2,4-diene-l-triphenyl phosphonium bromide was reacted with 3-formyl-crotonic acid butyl ester in presence of sodium hydride as base and dimethylformamide as solvent. The resultant ester derivative was obtained with a trans is (E/Z) ratio of around 55:45 which was subjected to hydrolysis in presence of potassium hydroxide and ethyl alcohol to obtain acitretin.
Use of hazardous, highly pyrophoric and moisture sensitive reagent like sodium hydride, along with cumbersome work-up and successive crystallizations to obtain the desired isomer rendered the process unviable for commercial scale.
Indian patent application 729/MUM/2012 discloses use of organic bases such as triethyl amine or pyridine for the reaction of 3-formyl-crotonic acid butyl ester and 5-(4-methoxy-2,3,6-trimethylphenyl)-3-methyl-penta-2,4-diene-l -triphenyl phosphonium bromide for the synthesis of acitretin. The process utilizes a large excess of the organic base (2.85:1.0) with respect to the reactant phosphonium bromide derivative. Further, there is no mention of the ratio of cis and trans geometric isomers of the product thus obtained either at the intermediate or final stage. The trans: cis (E/Z) ratio of the intermediate significantly impacts the final yield and purity of the product as several purifications and crystallizations are required to obtain the desired trans isomer.
The present inventors have experimentally observed that use of organic base in such large quantities severely hampers the removal of the undesired side product triphenyl phosphonium oxide formed in significant amounts. Also, the intermediate is obtained with a very modest trans: cis (E/Z) ratio.
WO2012/155796 discloses another method wherein alkali metal alkoxides are used as bases in the reaction of 5-(4-methoxy-2,3,6-trimethylphenyl)-3 -methyl -penta-2,4-diene-l -triphenyl phosphonium bromide with 3-formyl-crotonic acid. The obtained reaction mass, after adjusting pH to 7-8 with acid, is directly subjected to catalytic isomerization using catalysts such as Pd(OAc)2 or Pd(NH3)2Cl2. The reaction mixture so obtained is quenched with water, neutralized and filtered to get the desired product, which is further recrystallized from ethyl acetate. Although this procedure avoids the hydrolysis step and attempts in-situ isomerization, however the use of expensive, soluble palladium catalyst which cannot be recycled from the reaction mass coupled with lengthy reaction time of 25-30 hours and large solvent volumes make the process unviable.
It may be noted that in the synthesis of acitretin, the key reaction of 5-(4-methoxy-2,3,6-trimethylphenyl)-3 -methyl-penta-2 ,4-diene- 1 -triphenylphosphoniumbromide with 3 -formyl crotonic acid or its ester in presence of either strong inorganic bases such as sodium hydride, alkali metal alkoxides or organic bases like triethylamine is common to almost all synthetic routes disclosed in the prior art. Hence, all these routes suffer from the inherent problems of formation of undesired impurities including cis-isomeric compounds and their separation from the desired all-trans product which necessitates various purification methods ranging from column chromatography, multiple crystallizations etc.
Thus, there still exists a need for a convenient, easy-to-scale up process for synthesis of acitretin (I) which avoids use of pyrophoric strong bases and provides a robust method which affords acitretin having desired isomeric purity in high yield.
5-(4-methoxy,2,3,6 trimethylphenyl)- 3-formyl crotonic acid butyl glyoxalate L(+) tartaric acid
3-methyl-penta-2,4-dien-1-triphenyl butyl ester (V) dibutyl ester
phosphonium bromide (IV)
Acitretin (I)
Satish Mehta,CEO, Above and here Inspiring the participants
EXAMPLES
Example 1: Preparation of 4-(4-methoxy-2,3,6-trimethylphenyl)-but-3-en-2-one (II)
Acetone (6000 ml) was added to 4-methoxy-2,3,6 trimethyl benzaldehyde (500.3 g) and the mixture was stirred at 20-30°C. Aqueous solution of sodium hydroxide (134.8 g in 500 ml water) was gradually added to it and the resulting mixture was heated to 45-50°C with continued stirring. After completion of the reaction, as monitored by HPLC, the reaction mass was cooled and acetic acid was added till pH 4.5 to 5.5. Distillation of acetone, followed by addition of cyclohexane to the residue, followed by washing with water, separation and concentration of the organic layer gave 4-(4-methoxy-2,3,6 trimethylphenyl)-but-3-en-2-one of formula (II).
Yield: 80-84%
Example 2: Preparation of 5-(4-methoxy-2,3,6-trimethylphenyl)-3-methyl-penta-2,4-diene- 1-triphenyl phosphonium bromide (IV)
4-(4-Methoxy-2,3,6-trimethylphenyl)-but-3-en-2-one (II; 500 g) dissolved in toluene (2000 ml) was gradually added to a mixture of vinyl magnesium bromide (3500 ml; 1 molar solution in THF) and lithium chloride (4.8 g) and stirred at 20-30 C till completion of the reaction as monitored by HPLC. The reaction mixture was quenched with water and concentrated hydrochloric acid was added till the pH was between 3 and 4. The organic layer was separated and concentrated to give residue containing 5-(4-methoxy-2,3,6 trimethylphenyl)-3 -methyl -penta l,4-dien-3-ol (III). Methyl isobutyl ketone (3500 ml) was added to the residue, followed by gradual addition of triphenyl phosphine hydrobromide (745.3 g) at room temperature. The reaction mixture was heated to 50-60°C till completion of the reaction. The reaction mixture was cooled and filtered to give 5-(4-methoxy-2,3,6-trimethylphenyl)-3-methyl-penta-2,4-diene-l-triphenyl phosphonium bromide of formula (IV).
Yield: 1000 g (76%)
Example 3: Preparation of 3-formyl crotonic acid butyl ester (V)
Dibutyl-L- tartrate (500 g) was dissolved in isopropanol (3500 ml) at room temperature, and water (750 ml) was added to it. The reaction mixture was cooled to 15-25°C and sodium metaperiodate (448.5 g) was gradually added to it with stirring. The reaction was continued at 20-30°C till completion of the reaction based on GC analysis. The reaction mixture was filtered and the filtrate was concentrated. The resulting residue was dissolved in toluene (1000 ml), stirred and filtered to obtain the filtrate containing butyl glyoxylate. Propionaldehyde (221.0 g) was added to the filtrate and heated to around 60°C, followed by gradual addition of piperidine (26.4 g, dissolved in toluene). The reaction mixture was further heated and stirred at 110-120°C till completion of the reaction, as monitored by GC. After completion, the reaction mass was cooled, washed with aqueous sulfuric acid, water and finally with aqueous sodium bicarbonate solution. The organic layer was concentrated and the residue was distilled to give 3-formyl crotonic acid butyl ester (V)
Yield: 230-280 g (35-43%)
Example 4. Preparation of butyI{(2E,4E,6E,8E)-9-(4-methoxy-2,3,6-trimethyl) phenyl-3,7-dimethyl-nona-2,4,6,8}tetraenoate (VI)
Sodium carbonate (297. lg), was added to the mixture of 5-(4-Methoxy-2,3,6-trimethylphenyl)-3-methyl-penta-2,4-diene-l-triphenyl-phosphoniumbromide (IV; 1000 g) in toluene (5000 ml) followed by gradual addition of 3-formyl crotonic acid butyl ester (330 g) at room temperature. The stirred reaction mixture was heated to 60-70°C till completion of the reaction as monitored by HPLC. The reaction mass was cooled, quenched with water. The organic layer was separated, concentrated and n-heptane was added to the residue. The mass was stirred, filtered and 40% aqueous methanol (2000 ml) was added to it with stirring. Layer separation, concentration of the organic layer, and crystallization of the resulting residue from isopropyl alcohol, optionally with seeding followed by filtration gave crop I of butyl {{(2E,4E,6E,8E)— 9-(4-methoxy-2,3,6 trimethyl)phenyl-3,7 dimethyl -nona-2,4,6,8} tetraenoate (VI),.
Yield: 45-50%;
Cis: Trans isomer ratio (2.0:98.0)
The filtrate was concentrated, the residue was dissolved in toluene (2000 ml) and treated with iodine (4.5 g) at room temperature. After completion of the reaction, as monitored by HPLC, the reaction mixture was stirred with aqueous sodium thiosulfate solution. Separation and concentration of the organic layer and crystallization of the resulting residue from isopropyl alcohol, optionally with seeding, gave crop II of butyl {{(2E,4E,6E,8E)-9-(4-methoxy-2,3,6-trimethyl)phenyl-3,7-dimethyl-nona-2,4,6,8} tetraenoate (VI).
Yield (crop II): 15 to 20%.
Cis: Trans isomer ratio (2.0:98.0)
Total yield (crop I+II): 60-70%.
Example 5: Preparation of acitretin (I)
Aqueous solution of potassium hydroxide (155.2 g in 600 ml water) was added to a solution of butyl {(2E,4E,6E,8E)-9-(4-methoxy-2,3 ,6-trimethyl) phenyl-3 ,7-dimethyl-nona- 2,4,6,8}tetraenoate, VI (300.0 g) in ethanol (1800 ml) at 25-30°C and the reaction mixture was stirred at reflux temperature till completion of the reaction. After completion, as monitored by HPLC, the reaction mixture was quenched with water, and hydrochloric acid was added till pH was between 2.5 and 3.5. The mass was heated at 70°C, stirred, cooled to 40-50°C and filtered. Recrystallization of the resulting solid from tetrahydrofuran gave acitretin (I).
Yield: 154.0 g (60%)
Desired trans isomer: > 98%
India's hockey stars Sardara Singh and Sandeep Singh with Emcure Pharmaceuticals COO, Arun Khanna
 HE Dr. Kenneth Kaunda, First President of Zambia interacting with Mr. A. K. Khanna, COO & ED, Emcure at Emcure booth at AIDS 2012 conference, Washington
HE Dr. Kenneth Kaunda, First President of Zambia interacting with Mr. A. K. Khanna, COO & ED, Emcure at Emcure booth at AIDS 2012 conference, Washington
Mr. Sunil Mehta is an Executive Director and Senior Director (Projects)
Arun Khanna is the Chief Operating Officer and Executive Director on the Board of Emcure Pharmaceuticals Limited.
//////New patent, WO 2016042573, Acitretin, Emcure Pharmaceuticals Ltd