US 8362006
Intervet International B.V., Boxmeer, The Netherlands
Process for Making Zilpaterol and Salts Thereof
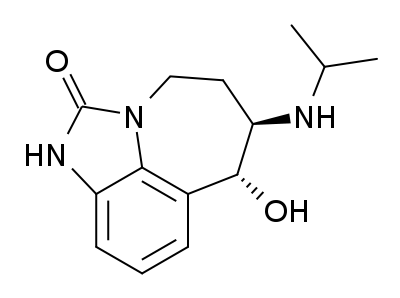
Zilpaterol is a known adrenergic β-2 agonist having the following structure:

The IUPAC name for zilpaterol is 4,5,6,7-tetrahydro-7-hydroxy-6-(isopropylamino)imidazo[4,5,1-jk]-[1]benzazepin-2(1H)-one. The Chemical Abstracts name for zilpaterol is 4,5,6,7-tetrahydro-7-hydroxy-6-[(1-methyl-ethyl) amino]-imidazo [4,5,1-jk][1]benzazepin-2(1H)-one.It is well known that zilpaterol, various zilpaterol derivatives, and various pharmaceutically acceptable acid addition salts of zilpaterol and its derivatives may, for example, be used to increase the rate of weight gain, improve feed efficiency (i.e., decrease the amount of feed per amount of weight gain), and/or increase carcass leanness (i.e., increase protein content in carcass soft tissue) in livestock, poultry, and/or fish. In U.S. Pat. No. 4,900,735, for example, Grandadam describes zootechnical compositions of racemic trans zilpaterol and salts thereof that may be used to increase the weight and meat quality of warm-blooded animals, including cattle, pigs, and poultry. And U.S. Patent Appl. Publ. US2005/0284380 describes use of an ionophore/macrolide/zilpaterol dosing regimen to increase beef production, reduce feed intake while maintaining beef production, and reduce incidences of liver abscess in cattle.
Methods for making zilpaterol are known in the art. For example, in U.S. Pat. No. 4,585,770, Fréchet et al. describe compounds encompassed by a genus characterized as 6-amino-7-hydroxy-4,5,6,7-tetrahydro-imidazo[4,5,1-jk][1]-benzazepin-2[1H]-one derivatives and pharmaceutically acceptable acid addition salts thereof. The derivatives correspond in structure to the following formula:
Here, R can be various substituents, and the wavy lines indicate that the bonds to the 6-amino and 7-OH groups have the trans configuration. This genus encompasses racemic trans zilpaterol when R is isopropyl.The methods reported in U.S. Pat. No. 4,585,770 use 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime as an intermediate. This compound corresponds in structure to the following formula:

As indicated in U.S. Pat. No. 4,585,770, 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime may be formed from starting materials that have been long known in the art. U.S. Pat. No. 4,585,770 illustrates the use of two such starting materials. In both examples, the starting materials are used to form 5,6-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,7-[1H,4H]-dione, which, in turn, may be used to make 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime.In one of the examples in U.S. Pat. No. 4,585,770, the starting material is 1,3-dihydro-1-(1-methylethenyl)-2H-benzimidazol-2-one, which is described in
J. Chem. Soc. Perkins, p. 261 (1982):
U.S. Pat. No. 4,585,770 indicates that 1,3-dihydro-1-(1-methylethenyl)-2H-benzimidazol-2-one may be reacted with an alkyl 4-halobutyrate (i.e., R
A—(CH
2)
3—COOR
B (wherein R
A is Cl, Br, or I; and R
B is C
1-C
4-alkyl), such as methyl or ethyl 4-bromobutyrate) and a base (e.g., an alkali metal) to form a butanoate, which, in turn may be hydrolyzed with an acid (e.g., H
2SO
4) in an alkanol (e.g., methanol or ethanol) to remove the methylethenyl substituent. The hydrolysis product then may be subjected to saponification by reacting it with a base (e.g., NaOH or KOH) in an alkanol to form a carboxylic acid. Subsequently, the carboxylic-acid-terminated side chain may be cyclized to form 5,6-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,7-[1H,4H]-dione by reacting the carboxylic acid with thionyl chloride to obtain a chloride, and then treating the chloride with a Lewis acid (e.g., aluminum chloride) in an organic solvent (e.g., methylene chloride or dichloroethane):
See U.S. Pat. No. 4,585,770, col. 4, line 3 to col. 5, line 14; and Example 14, col. 12, lines 1-68.In another example in U.S. Pat. No. 4,585,770, the starting material is 1,3-dihydro-1-benzyl-2H-benzimidazol-2-one, which is described in
Helv., Vol 44, p. 1278 (1961):

U.S. Pat. No. 4,585,770 indicates that the 1,3-dihydro-1-benzyl-2H-benzimidazol-2-one may be reacted with ethyl 4-bromobutyrate and sodium hydride to form 1,3-dihydro-2-oxo-3-benzyl-1H-benzimidazol-1-butanoate, which, in turn may be subjected to saponification by reacting it with methanolic NaOH to form 1,3-dihydro-2-oxo-3-benzyl-1H-benzimidazol-1-butanoic acid. The butanoic acid side chain may then be cyclized by reacting the 1,3-dihydro-2-oxo-3-benzyl-1H-benzimidazol-1-butanoic acid with thionyl chloride to obtain a chloride, and then treating the chloride with aluminum chloride in dichloroethane. The cyclized product, in turn, may be hydrolyzed using o-phosphoric acid in phenol to form 5,6-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,7-[1H,4H]-dione. See U.S. Pat. No. 4,585,770, Example 1, Steps A-D, col. 6, line 10 to col. 7, line 35.Using the methods reported in U.S. Pat. No. 4,585,770, 5,6-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,7-[1H,4H]-dione may be reacted with an alkyl nitrite (e.g., tert-butyl nitrite or isoamyl nitrite), in the presence of a base or acid (e.g., HCl), to form 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime. The 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime, in turn, is reduced via catalytic hydrogenation (with, for example, hydrogen in the presence of palladium on carbon) or sodium borohydride to form racemic trans 6-amino-7-hydroxy-4,5,6,7-tetrahydro-imidazo[4,5,1-jk][1]-benzazepin-2[1H]-one:

In the illustrative example in U.S. Pat. No. 4,585,770, the 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime is converted into racemic trans 6-amino-7-hydroxy-4,5,6,7-tetrahydro-imidazo[4,5,1-jk][1]-benzazepin-2[1H]-one in two steps: the 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime is first reacted with H
2 in the presence of Pd-on-carbon, and, then, after filtration, the hydrogenation product is reacted with sodium borohydride. See U.S. Pat. No. 4,585,770, col. 2, line 15 to col. 4, line 2; and Example 1, Steps E & F, col. 7, line 38 to col. 8, line 3.U.S. Pat. No. 4,585,770 reports that the trans stereoisomers of 6-amino-7-hydroxy-4,5,6,7-tetrahydro-imidazo[4,5,1-jk][1]-benzazepin-2[1H]-one may be alkylated with acetone in the presence of a reducing agent (e.g., an alkali metal borohydride or cyanoborohydride, such as sodium cyanoborohydride) to form racemic trans zilpaterol:
See U.S. Pat. No. 4,585,770, col. 2, line 46 to col. 4, line 2; and Example 13, col. 11, lines 41-68.In view of the importance of zilpaterol and its salts in animal production, there continues to be a need for cost-effective, high-yield processes for making zilpaterol and its salts. The following disclosure addresses this need.
OVERVIEW
Zilpaterol 121 is used to increase the rate of weight gain in livestock, poultry, and fish. The drug is available as Zilmax and is marketed as beef improvement technology. There are a number of methods for preparing 121, and the patent specifically focuses on the method reported in a 1986 patent, U.S. 4,585,770, that is compared with the process described in the current patent.
The new process is outlined in Schemes 37 and 38, and the examples in the patent describe the manufacture of 121 on a commercial scale starting from 525 kg of 116a.
Unfortunately, the yield of the reaction products is not reported in any of the steps. The process starts with the chlorination of the acid 116a to give 116b that is carried out using (COCl)2, although COCl2 or triphosgene are also claimed to be suitable. The product is isolated as a solution in DCM after a workup involving transferring between three vessels, adding H2O, and distilling off the solvent.
In the next stage an intramolecular Friedel–Crafts alkylation of 116b in the presence of AlCl3 followed by acid hydrolysis forms 117. This is isolated as a wet solid and then is converted to the oxime 118a in DMF by treatment with NaNO2 followed by addition of HCl.
Compound 118a is isolated as a dry solid that is converted to the potassium salt by treatment with 45% aq KOH as shown in Scheme 38. The salt is isolated as a solution that is treated with active C and then hydrogenated in the presence of Pd/C catalyst to form the amino alcohol salt 119.
This reaction appears to be stereoselective, although no reference to this is made in the patent. The salt, 119, is recovered as an aqueous solution that is used in the next step where it is reacted with Me2CO in the presence of HOAc at a pH of 7–8. This produces the isopropylidene amino compound, 120, that is not isolated but undergoes hydrogenation in the presence of Pt/C catalyst to give the HOAc salt, 121·HOAc.
The free base form, 121, is obtained by treating the salt with NaOH in EtOH, and from the free base, a HCl salt can be prepared.
The patent discusses aspects of the process is some detail such as the quantities of washing solvents used.
Advantages
The process provides an effective route to the desired compound and is clearly suitable for large-scale manufacture.
The following Scheme I generically illustrates a scenario wherein all the above reactions are used:
The following Scheme II generically illustrates the above scenario wherein the chlorinating agent comprises oxalyl chloride; the Lewis acid comprises AlCl3; the hydrolysis acid following the Friedel-Crafts reaction comprises HCl; the inorganic nitrite comprises NaNO2; the acid used in the oximation comprises HCl; water is added to the oximation product mixture to foster isolation of the oxime product; the base used to form the oxime salt comprises KOH; the catalyst for the first hydrogenation comprises palladium on carbon; the acid used in the formation of the isopropylideneamino compound comprises acetic acid; the catalyst for the second hydrogenation comprises platinum on carbon; and the base and alcohol used to form the zilpaterol free base comprise NaOH and ethanol, respectively:

Example 1 Preparation of 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione Part A. Preparation of chloro 2,3-dihydro-2-oxo-1H-benzimidazol-1-butanoate
4-(2-Oxo-2,3-dihydrobenzimidazol-1-yl)butyric acid (50 g; 0.227 mol), N,N-dimethylformamide (1.84 g; 0.025 mol; 0.11 eq), and dichloromethane (480 g; 5,652 mol; 24.89 eq) were charged to a stirred-tank reactor. Oxalyl chloride (31.12 g; 0.245 mol; 1.08 eq) was then dosed at 10-20° C. over a 1-hour period while stirring. The resulting mixture was then stirred at 10-20° C. for an additional hour. All the above steps were conducted under a N
2 atmosphere.Part B. Preparation of 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione.

The reaction product mixture from Part A was added to a slurry of aluminum chloride (100 g; 0.75 mol, 3.3 eq) in dichloromethane (320 g; 3.768 mol; 16.59 eq) over 2-5 hours at 60° C. and a pressure of 2.7 bar (absolute) in a stirred-tank reactor that allowed HCl gas to escape through an overpressure vent. The resulting slurry was stirred for an additional hour at that temperature, and then cooled to 12° C. In a separate stirred-tank reactor, water (800 g; 44.407 mol; 195.59 eq.) and aqueous 32.5% HCl (118 g; 1.052 mol HCl; 4.63 eq. HCl) were mixed. This mixture was cooled to 0° C., and the gas in the headspace was evacuated to 300 mbar (absolute). The slurry from the first reactor was then added portion-wise to the second reactor, whereby the temperature increased to 10-15° C. under distillation of dichloromethane. The first reactor was rinsed with additional dichloromethane (25 g; 0.294 mol; 1.3 eq), which was then added to the second reactor. Distillation of the dichloromethane was then completed at 300 mbar to atmospheric pressure (absolute) and 12-40° C. The resulting suspension was cooled to 0° C. The solid was filtered off, and washed 4 times with water (291.25 g each time; 64.668 mol total; 284.83 eq. total) and once with isopropanol (80 g; 1.331 mol; 1.331 eq) at 0° C. All the above steps were conducted under a N
2 atmosphere.Example 2 Preparation of 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime.

8,9-Dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione (50 g; 92.4% purity; 0.228 mol) prepared in accordance with the procedure in Example 1 was dried and mixed with isopropanol (7.23 g; 0.12 mol; 0.53 eq) and water (3.01 g; 0.167 mol; 0.73 eq) (in alternative experiments and in production, 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione prepared in accordance with the procedure in Example 1 was instead used as centrifuge-wet material without the addition of water and isopropanol). The resulting wet 8,9-dihydro-2H-7H-2,9a-diazabenzo[cd]azulene-1,6-dione was combined with sodium nitrite (19.05 g at 99.3% purity; 0.274 mol; 1.2 eq) and N,N-dimethylformamide (800 g; 10.945 mol; 47.9 eq) in a stirred-tank reactor. The mixture was heated to 50° C., and then 32% HCl (41.65 g; 0.366 mol HCl; 1.6 eq HCl) was added over a 30 minute period. Toward the end of the HCl addition (i.e., after greater than 1 eq HCl had been added), the temperature quickly increased to 60-70° C. After all the HCl was added, the mixture was stirred at 60° C. for an additional 30 minutes. The mixture then was cooled to 35° C. over a 2- hour period. Next, water (224.71 g; 12.473 mol; 54.6 eq) was added over a 2-hour period. The resulting mixture was then cooled to 0° C. over a 2-hour period, and maintained at that temperature for 2 hours. Afterward, the solid 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime product was removed by filtration and washed 4 times with water (70.1 ml each time; 15.566 mol total; 68.13 eq total) and once with acetone (115.9 g; 99.9% purity; 1.994 mol; 8.73 eq). All the above steps were conducted under a N
2 atmosphere.Example 3 Scale-up Preparation of 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione Part A. Preparation of chloro 2,3-dihydro-2-oxo-1H-benzimidazol-1-butanoate
Dichloromethane (3772 L) and then 4-(2-oxo-2,3-dihydrobenzimidazol-1-yl)butyric acid (525 kg; 2.4 kmol) were charged to a stirred-tank reactor, followed by N,N-dimethylformamide (21 L). The resulting mixture was cooled to 10° C. Afterward, oxalyl chloride (326.8 kg)) was dosed at 10-15° C. over 2-3 hours while stirring. The resulting mixture was then stirred at 15-20° C. for an additional 1-3 hours. All the above steps were conducted under a N
2 atmosphere. Conversion was checked by in-process control (“IPC”).Part B. Preparation of 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione.
Aluminum chloride (1050 kg) and dichloromethane (2403 L) at 10-20° C. were charged to a stirred-tank reactor, followed by additional dichloromethane (112 L) at 10-20° C. to rinse the reactor. The reactor was then pressurized with N
2 to 2.7 bar (absolute), and heated to 58-60° C. Next, the product mixture from Part A was added over 2−5 hours. The resulting slurry was stirred for an additional 1-2 hours, and then cooled to 10-20° C. Afterward, the pressure was released. In a second stirred-tank reactor at 5° C., water (3675 L) was charged, followed by aqueous 33% HCl (452 L). This mixture was cooled to 0° C., and the gas in the headspace was evacuated to 270-470 mbar (absolute). About half the content from the first reactor was added to the second reactor at from 5-20° C. The mixture was maintained at 10-30° C. for an additional 30-90 minutes. In parallel to and following the transfer, distillation of dichloromethane occurred. The line between the two reactors was rinsed with dichloromethane (150 ml). The resulting rinse and the contents in the second reactor were transferred to a thud stirred-tank reactor. The transfer line between the second and third reactors was rinsed with water (200 L). This rinse also was charged to the third reactor. Water (3675 L) at 5° C. and 33% HCl (452 L) were then added to the second reactor. The resulting mixture was cooled to 0° C., and the pressure in the headspace was set to between 270-470 mbar (absolute). The second half of the content from the first reactor was then added to the second reactor at 5-20° C. This mixture was maintained at 10-30° C. for an additional 30-90 minutes. In parallel to and following the transfer, distillation of dichloromethane occurred. The line between the first and second reactors was rinsed with dichloromethane (150 ml). The resulting rinse and the contents in the second reactor were transferred to the third reactor. The transfer line between the second and third reactors was then rinsed with water (200 L). This rinse was charged to the third reactor. In the third reactor, the dichloromethane was further distilled at 30-40° C. under atmospheric pressure. When the distillation was complete, the suspension was cooled to 0−5° C., and then centrifuged in two parts. Each of the resulting cakes was washed with four times water (390 L for each wash) and once with isopropanol (508 L) at 0−5° C. All the above steps were conducted under a N
2 atmosphere.Example 4 Scale-up of Preparation of 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime.

At 20° C., N,N-dimethylformamide (7068 L) was charged to a stirred-tank reactor, followed by 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione (450 kg total wet material, approximately 405 kg pure) prepared in accordance with the procedure in Example 3. The addition funnel was rinsed with N,N-dimethylformamide (105 L), and the rinse was charged to the reactor. The resulting mixture was heated at 45° C. until all the 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione was in solution. IPC was used to check the amount of pure 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione in the mixture, and, from that measurement (together with the mass of wet 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione and N,N-dimethylformamide), the exact amount of 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione was calculated, which, in turn, was used to calculate the amounts of N,N-dimethylformamide (17.3 kg/kg), sodium nitrite (0.412 kg/kg) and HCl 33% (0.873 kg/kg). For the duration of the IPC, the mixture was cooled to 20° C. Next, sodium nitrite (167 kg, based on 405 kg 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione) was added. The addition funnel was rinsed with N,N-dimethylformamide (105 L), and the rinse was charged to the reactor. The temperature was then increased to 45° C. Subsequently, additional N,N-dimethylformamide was charged in the amount calculated earlier (97 L, based on having a total of 7375 L DMF for 405 kg of 8,9-dihydro-2H,7H-2,9a-diazabenzo[cd]azulene-1,6-dione). Next, the resulting mixture was warmed to 48° C., and then 33% HCl (353 kg, based on the batch size) was added over 1 hour, causing the temperature to increase to 60-65° C. by the end of the addition. The mixture was then stirred at 60° C. for another 30 minutes. Next, the mixture was cooled to 45° C. over 1-2 hours. The resulting mixture was transferred into a second reactor. The first reactor was subsequently rinsed with N,N-dimethylformamide (105 L), and the rinse was charged to the second reactor. Water (2000 L) was then added over a 2-hour period at 38° C. The resulting mixture was cooled to 0° C. over 2-3 hours, and then stirred at that temperature for another 2-8 hours. Afterward, the mixture was centrifuged at 0° C., and the resulting cake was washed with three times with water (810 L each time), washed with acetone (1010 L), and dried at 65° C. under vacuum. All the above steps, except for the IPC, were conducted under a N
2 atmosphere.Example 5 Preparation of Zilpaterol Part A. Formation of Aminoalcohol Potassium Salt from Ketooxime
A stirred-tank reactor was purged 3 times with N
2 between high pressure (3 bar, absolute) and low pressure (1 bar, absolute) for 10 minutes each. Then a pressure of 0.9 bar (absolute) was established. Water (790 kg) was then charged to the reactor, followed by 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime (255 kg) prepared in accordance with Example 4. The reactor contents were then heated to 40° C. Next, 45% KOH (214 kg) was continuously charged to the reactor, causing 4,5-dihydro-imidazo[4,5,1-jk][1]benzazepin-2,6,7[1H]-trione-6-oxime to form the corresponding potassium salt, which, in turn, dissolved (this could be visually verified). The reactor was then charged with active charcoal (13 kg). The resulting mixture was then stirred for 30 minutes at 40° C. The resulting mixture was filtered through a filter loop for one hour to remove the active charcoal. The mixture was then cooled to 15° C. A 5% palladium-on-carbon catalyst (25.5 kg, Johnson-Matthey) was then charged to the reactor. The reactor was then rinsed with water (50 kg). The resulting mixture in the reactor was stirred for 2-6 hours at 40° C. and a H
2 pressure of 5-10 bar (absolute). Afterward, the reactor was vented over 30 minutes, and the reaction was analyzed using HPLC. The contents were then filtered in a filter loop for 90 minutes. The filter cake was washed with water (50 L), and removed to recover palladium. The filtered solution was analyzed via HPLC to confirm complete conversion, and then used in the next step.Part B. Formation of zilpaterol-HOAc.
The solution from Part A was cooled to 30° C. Acetone (625 L) was then charged to the reactor. Acetic acid was added to adjust the pH to 7.5 (a pH of from about 7 to about 8 is preferred). The resulting mixture was then cooled to 15° C. Next, a 5% platinum-on-carbon catalyst (21.3 kg, Degussa) was charged to the reactor, followed by water (50 kg) to rinse the reactor. The head space was purged 3 times with H
2 between a high pressure of 5 bar (absolute) and a low pressure of 1 bar (absolute) for 15 minutes each. Then a hydrogen pressure of 9.0 bar (absolute, for hydrogenation) was established. The mixture was heated to 70° C. over 1 hour while being stirred, and then maintained at that temperature for an additional hour while being stirred. The reactor was then vented, and the headspace was purged with N
2. The reaction was analyzed using HPLC. Acetic acid (8 kg) was then charged to the reactor, and the resulting mixture was cooled to 30° C. More acetic acid was added to adjust the pH to 6.8. The mixture was then transferred through a filter loop for 1 hour while being maintained at 30° C. The resulting cake was washed with 7% aqueous acetic acid (75 L). The filtered solution was transferred to another stirred-tank reactor to be used in the next step.Part C. Formation of Zilpaterol Free Base
The stirred-tank reactor containing the product from Part B was purged 3 times with N
2 between high pressure (2 bar, absolute) and low pressure (1 bar, absolute) for 10 minutes each. Then a pressure of 0.9 bar (absolute) was established. Next, the mixture was concentrated by distillation to 30-70%. The concentrated mixture was cooled to 65° C. Ethanol (331 L) was charged to the reactor, and the resulting mixture was cooled to 50° C. The pH was adjusted to 10 using 25% NaOH. This caused zilpaterol free base to precipitate. The temperature was decreased to 0° C. to facilitate the precipitation, and maintained at that temperature for an additional hour. The solids were filtered off, and washed with water (700 L).Example 6 Synthesis of an HCl Salt of the ZilpaterolThe free base of zilpaterol is dissolved in ethanol. Subsequently, ethyl acetate saturated with HCl is added. The resulting mixture is vacuum-filtered to obtain a crude product containing the HCl salt of the zilpaterol. The crude product is dissolved in hot methanol. Ethyl acetate is then added, and the mixture is filtered to obtain the final HCl salt product.
Example 7 First Illustration of a Contemplated Suitable Dosage FormA tablet is prepared containing 2.5 or 5 mg of the HCl salt of Example 6, and sufficient excipient of lactose, wheat starch, treated starch, rice starch, talc, and magnesium stearate for a final weight of 100 mg.
Example 8 Second Illustration of a Contemplated Suitable Dosage FormGranules are prepared containing 12.5 or 25 of the HCl salt of Example 6 in each daily dose of granules.
Example 9 Third Illustration of a Contemplated Suitable Dosage FormThe HCl salt of Example 6 is crystallized using the methodology discussed U.S. Pat. No. 5,731,028 for making crystalline racemic trans zilpaterol. Less than 5% of the crystals have a size of less than 15 μm, and at least 95% of the crystals have a size of less than 250 μm. A premix of the crystalline HCl salt secured to a 300-800 μm corn cob support is then obtained using the methodology discussed in European Patent 0197188 (incorporated by reference into this patent). The concentration of the HCl salt in the premix is 3% (by weight).
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|
| US4585770 | 12 Oct 1983 | 29 Apr 1986 | Roussel Uclaf | Novel 6-amino-7-hydroxy-4,5,6,7-tetrahydro-imidazo[4,5,1-j-k][1]-benzazepin-2-(1H)-one |
| US5731028 | 6 Jun 1996 | 24 Mar 1998 | Roussel Uclaf | Crystallized zilpaterol hydrochloride |
| US20060040950 | 17 Dec 2003 | 23 Feb 2006 | Janssens Frans E | Substituted 1-piperidin-4-yl-4-pyrrolidin-3-yl-piperazine derivatives and their use as neurokinin antagonists |
| US20080267942 * | 11 Apr 2008 | 30 Oct 2008 | Pfizer Limited | Benzazepin-2(1h)-one derivatives |
| US20100173892 * | 31 Jan 2008 | 8 Jul 2010 | Juan Jose Almena-Perea | Enantioselective synthesis of 6-amino-7-hydroxy-4,5,6,7-tetrahydro-imidazo[4,5,1-JK][1]-benzazepin-2[1H]-one and zilpaterol |
| WO2004056799A2 | 17 Dec 2003 | 8 Jul 2004 | Janssen Pharmaceutica N.V. | Substituted 1-piperidin-4-yl-4-pyrrolidin-3-yl-piperazine derivatives and their use as neurokinin antagonists |
| WO2008119754A1 | 28 Mar 2008 | 9 Oct 2008 | Intervet International B.V. | Processes for making zilpaterol and salts thereof |
////////US 8362006, Intervet International B.V., Boxmeer, The Netherlands, Zilpaterol, PATENT