WO 2016025720, New Patent, by Assia Chemicals and Teva on Ibrutinib
ASSIA CHEMICAL INDUSTRIES LTD. [IL/IL]; 2 Denmark Street 49517 Petach Tikva (IL)
TEVA PHARMACEUTICALS USA, INC. [US/US]; 1090 Horsham Road P.O. Box 1090 North Wales, PA 19454 (US)
TEVA PHARMACEUTICALS USA, INC. [US/US]; 1090 Horsham Road P.O. Box 1090 North Wales, PA 19454 (US)
COHEN, Meital; (IL).
COHEN, Yuval; (IL).
MITTELMAN, Ariel; (IL).
MOHA-LERMAN, Elana, Ben; (IL).
TZANANI, Idit; (IL).
LEVENFELD, Leonid; (IL)
COHEN, Yuval; (IL).
MITTELMAN, Ariel; (IL).
MOHA-LERMAN, Elana, Ben; (IL).
TZANANI, Idit; (IL).
LEVENFELD, Leonid; (IL)
The present invention encompasses solid state forms of Ibrutinib, including forms G, J and K, and pharmaceutical compositions thereof.
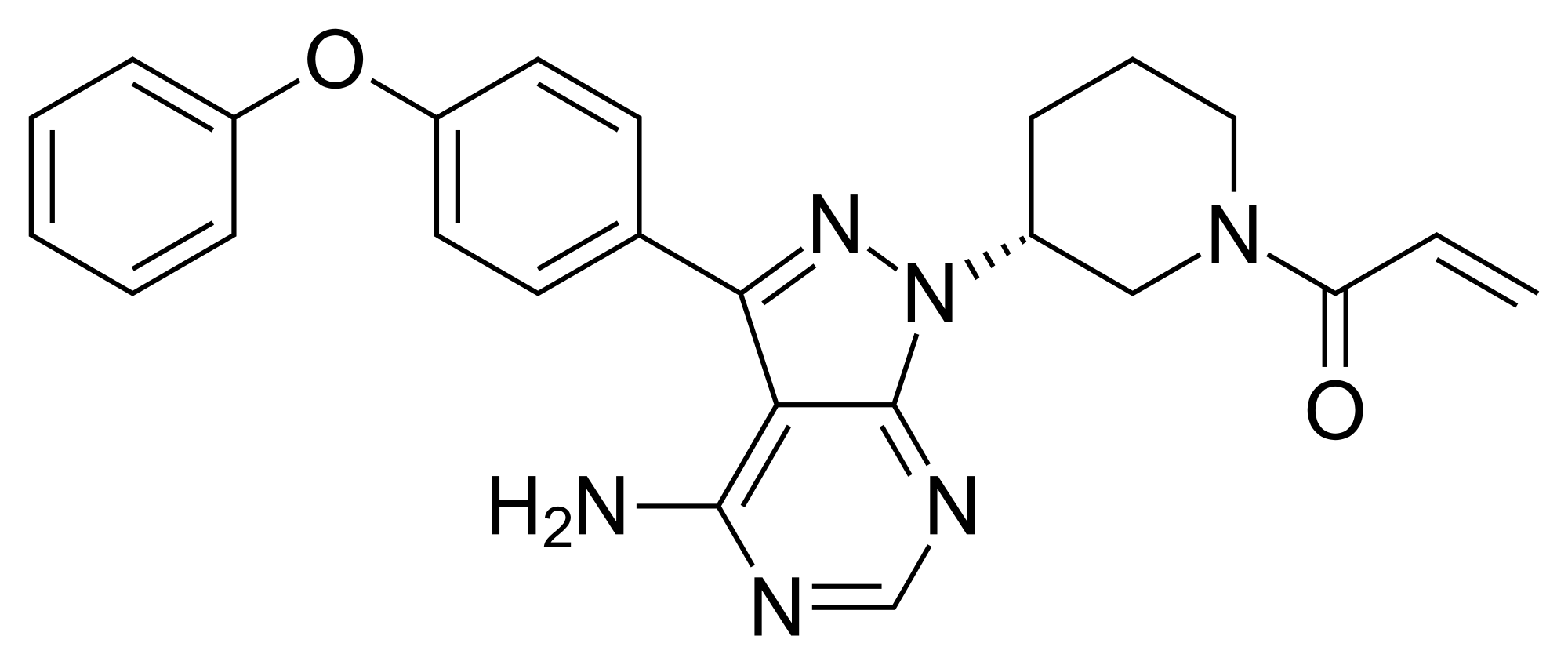
Ibrutinib, l-{(3R)-3- [4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo [3,4-d] pyrimidin-l-yl] piperidin-l-yl] prop-2-en-l-one, having the following formula,

is a kinase inhibitor indicated for the treatment of patients with B-cell lymphoma.
Ibrutinib is described in US 7,514,444 and in US 8,008,309. Solid state forms, including forms A-F and amorphous form of Ibrutinib, are described in WO 2013/184572.
Polymorphism, the occurrence of different crystalline forms, is a property of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis - "TGA", or differential scanning calorimetry - "DSC"), X-ray diffraction pattern, infrared absorption fingerprint, and solid state (13C-) NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Different salts and solid state forms (including solvated forms) of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms and solvates may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also offer improvements to the final dosage form, for instance, if they serve to improve bioavailability. Different salts and solid state forms and solvates of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
Discovering new solid state forms and solvates of a pharmaceutical product may yield materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification, or may serve as desirable intermediate crystal forms that facilitate purification or conversion to other polymorphic forms. New solid state forms of a pharmaceutically useful compound can also provide an opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, e.g., a different crystal habit, higher crystallinity or polymorphic stability which may offer better processing or handling characteristics, improved dissolution profile, or improved shelf-life (chemical/physical stability). For at least these reasons, there is a need for additional solid state forms (including solvated forms) of ibrutinib.
Example 1: Preparation of Crystalline Form G of Ibrutinib
[0057] Ibrutinib (0.3 gr, amorphous form) was dissolved in acetic acid (1.2 ml) and the obtained solution was stirred at room temperature overnight followed by the addition of water (2.4 ml). A gum was obtained which was turned into cloudy solution upon stirring. The obtained cloudy solution was stirred for 9 days at room temperature and the obtained precipitate was collected by suction filtration. The obtained solid was dried in an oven at 40°C under vacuum for 16h to obtain form G of Ibrutinib (0.12g), as confirmed by XRPD.
Example 2: Preparation of Crystalline Form J of Ibrutinib
Ibrutinib (5.2 g) was dissolved in Anisole (15 ml), the solution was stirred at room temperature until precipitation was occurred. The slurry was stirred over night at room temperature and the precipitate was collected by suction filtration. The cake was dried in a vacuum oven at 50°C overnight. The obtained product was analyzed by XRPD and found to be form J.
Example 3: Preparation of Crystalline Form J of Ibrutinib
Ibrutinib (10.5 g) was dissolved in Anisole (21 ml) and MTBE (32 ml), the solution was stirred at room temperature until precipitation was occurred . The slurry was heated to reflux and was gradually cooled to room temperature. After 3 hours the precipitate was collected by suction filtration. The obtained product was analyzed by XRPD and found to be form J.
Example 4: Preparation of Crystalline Form G of Ibrutinib
A I L reactor was charged with Ibrutinib (100 g), acetonitrile (417.5 ml_), water (417.5 ml_) and acetic acid (27.15 g). The mixture was heated to 90°C until dissolution; the solution was gradually cooled to 0°C, then heated to 25°C and stirred over 48 hours at 25°C. The obtained slurry was filtered and washed with water (100 ml_). The product was dried overnight in a vacuum oven at 40°C to obtain Ibrutinib form G (72.9 g), as confirmed by XRPD.
Example 5: Preparation of Crystalline Form G of Ibrutinib
A 250 mL round flask was charged with isopropanol (10 ml_) and water (120 ml_), and a solution of Ibrutinib (10 g) in Acetic acid (40 mL) was added dropwise. The mixture was stirred at 25°C for 48 hours. The obtained slurry was filtered and the wet product was slurried in water (50 mL) for 5 min and filtered again. The obtained product was dried under vacuum at room temp in the presence of a N2 atmosphere and found to be form G, as confirmed by XRPD.
Example 6: Preparation of Crystalline Form K of Ibrutinib
Ibrutinib (10 g) was dissolved in toluene (50 mL) and dimethylformamide (DMA) (30 mL) at room temperature, the solution was heated to 50 °C and water (30 mL) was added. The phases were separated and methyl tert-butyl ether (MTBE) (30 mL) was added to the organic phase. The solution was cooled in an ice bath and seeded with amorphous Ibrutinib. After further stirring at the same temperature the obtained slurry was filtered under vacuum. The obtained solid was analyzed by XRPD and found to be Form K (Figure 5).

//////////////WO 2016025720, WO-2016025720, New Patent, Assia Chemicals, Teva, Ibrutinib