
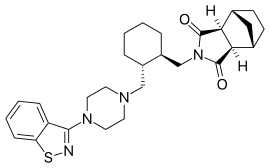
Carbotegravir, New Patent, WO 2016113372, Lek Pharmaceutical and Chemical Co DD
LEK PHARMACEUTICALS D.D. [SI/SI]; Verovskova 57 1526 Ljubljana (SI)
MARAS, Nenad; (SI).
SELIC, Lovro; (SI).
CUSAK, Anja; (SI)
SELIC, Lovro; (SI).
CUSAK, Anja; (SI)
ViiV Healthcare is developing cabotegravir (first disclosed in WO2006088173), which in July 2016, was reported to be in phase 2 clinical development.
WO-2016113372
Process for preparing integrase inhibitors such as dolutegravir and cabotegravir and their analogs, useful for treating viral infections eg HIV infection. Also claims a process for preparing intermediates of dolutegravir and cabotegravir.
(4R, 12aS)-N-[(2,4-Difluorophenyl)methyl]-3 ,4,6,8, 12, 12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-2H-pyrido[1 ',2':4,5]pyrazino[2, 1-b][1 ,3]oxazine-9-carboxamide (Formula A):

Formula A
known by the INN name dolutegravir, is a new efficient antiviral agent from the group of HIV integrase inhibitors which is used in combination with some other antiviral agents for treatment of HIV infections, such as AIDS. The compound, which belongs to condensed polycyclic pyridines and was first disclosed in WO2006/1 16764, is marketed.
Another compound disclosed in WO2006/1 16764 is (3S, 1 1 aR)-N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 1 1 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide (Formula

Formula C
known by the INN name cabotegravir.
The complex structures of dolutegravir and cabotegravir present a synthetic challenge. The first description of the synthesis in WO2006/1 16764 shows a 16-steps synthesis (see Scheme A), which is industrially impractical due to its length and low overall yield.

Scheme A
WO 2010/068253 and WO 2006/1 16764 describe an alternative synthesis. The 1 1 -step synthesis, shown in Scheme B1 and Scheme B2, is based on bromination of the 9-position for further introduction of the carboxylic group. The synthesis relies on the use of expensive palladium catalysts and toxic selenium compounds. Furthermore, some variations of these approaches involve pyrone intermediates in several steps. In some cases pyrones are liquids which can complicate purification, while further reactions form complex mixtures.

 doiutegravir
doiutegravir
Scheme B2
In further alternative syntheses, acetoacetates were used as starting materials. Such an approach is challenging in terms of introducing the hydroxy group in the 7-position. The variation in Scheme C1 , described in WO2012/018065, starts from 4-benzyloxyacetoacetate. The procedure requires 9 steps, but use expensive reagents like palladium catalysts. Moreover, there is described a possibility of formation a co-crystal between an intermediate and hydroquinone, wherein however the additional step may diminish yields and make the process longer and time consuming.

Scheme C1
The variation in Scheme C2, described in WO2012/018065, starts from 4-chloroacetoacetate. The process is not optimal because of problems in steps which include pyrones and because of problems with conversion of 7-chloro to 7-hydroxy group which includes a disadvantageous use of silanolates with low yield (25%).

Scheme C2
The variation in Scheme C3, described in WO201 1/1 19566, starts from unsubstituted acetoacetate. For the introduction of the 7-hydroxy group, bromination is used and substitution of bromo with hydroxy is performed by a use of silanolates. The substitution of the bromine is achieved in a 43% yield.

Scheme C3
The variation in Scheme C4, described in WO201 1/1 19566, starts from 4-methoxyacetoacetate aiming at preparing dolutegravir or cabotegravir. The process uses lithium bases to affect a difficult to control selective monohydrolysis of a diester.

The object of the present invention is to provide short, simple, cost-effective, environmentally friendly and industrially suitable processes for beneficially providing dolutegravir and analogues thereof and cabotegravir and analogues thereof, in particular dolutegravir.
Scheme 1

According to an embodiment of the process of the invention the building block 3-aminobutanol can suitably be substituted with other aminoalcohols to give dolutegravir analogues. For example, using (S)-alaninol gives cabotegravir as the final product. Similarly, using amines other than 2,4-difluorobenzylamine in the amidation step results in the synthesis of other dolutegravir analogues.
According to the another preferred embodiment cabotegravir or a pharmaceutically acceptable salt thereof is prepared by the analogue process, which comprises providing a compound of formula (5c)

5c
converting the compound of formula (5c) to a compound of formula (6c)

6c
by carrying out a chlorination reaction, and converting the compound of formula (6c) to cabotegravir and/or a pharmaceutically acceptable salt thereof.
The compound of formula (5c) can preferably be provided by converting a compound of formula (3) to a compound of formula (4c)

Scheme 2

1. ) EtOCOCI, Et3N / Me2CO
2. ) 2,4-difiuorobenzylamine

Scheme 3

Analogous compound of formula 7c is a useful intermediate in the synthesis of cabotegravir. Scheme 3a

Scheme 4

Examples
The following examples are merely illustrative of the present invention and they should not be considered as limiting the scope of the invention in any way. The examples and modifications or other equivalents thereof will become apparent to those versed in the art in the light of the present entire disclosure. Particularly, all Examples related to the preparation of dolutegravir and intermediates thereof can be used by the analogy for the preparation of cabotegravir and intermediates thereof.
Example 1 :

Methyl acetoacetate (1 , 25.22 g) and dimethylformamide dimethyl acetal (DMFDMA, 35 mL) was heated at 50-55°C for 2 h, then methanol (60 mL), aminoacetaldehyde dimethyl acetal (24 mL) and acetic acid (4 mL) was added an the mixture was heated under reflux for one hour, then concentrated. MTBE (100 mL) was added and the mixture was kept at 5 °C overnight to crystallize. Upon filtration 46 g (92%) of product 2 was recovered.
1H NMR (DMSO-d6): δ 2.31 (s, 3H), 3.30 (s, 6H), 3.49 (m, 2H), 3.61 (s, 3H), 4.43 (m, 1 H), 8.02 (d, 1 H), 10.8 (bs, 1 H). 13C NMR (DMSO-d6): δ 30.52, 35.48, 50.53, 54.23, 98.99, 102.47, 160.70, 166.92, 197.21 .
Example 2:

Compound 2 (5.00 g) was dissolved in 2-propanol, dimethyl oxalate (7.02 g) was added and heated to 40 °C. Sodium methylate (25% in methanol; 20 mL) was slowly (10 min) added, the mixture was then heated to 50-55 °C and stirred at that temperature for 2-2.5 h. The mixture was cooled to ambient temperature, then sodium hydroxide solution (1 M, 65 mL) was added to the mixture and stirred for another 2 h, followed by addition of concentrated hydrochloric acid (1 1 mL) and stirred for another 2 h. The precipitate was filtered and dried to give 8.08 g (NMR assay 47%; 65% yield) of compound 3.
1H NMR (DMSO-d6): δ 2.50 (m, 2H), 3.30 (s. 6H), 4.49 (m, 1 H), 7.06 (s, 1 H); 8.70 (s, 1 H). 13C NMR (DMSO-d6): δ 55.23, 55.37, 102.34, 1 15.47, 120.24, 145.17, 162.71 , 165.22, 178.55.
Example 3:

Compound 2 (158.37 g) was dissolved in methanol (548 mL), followed by the addition of dimethyl oxalate (202.2 g). While keeping the temperature below 30°C, potassium ferf-butoxide (192.1 g) was added and reaction mixture was heated at 50 °C overnight. The suspension was then filtered and the filter cake washed with methanol. The filtrate was concentrated (approximately to 680 mL), then water (680 mL) was added, followed by addition of lithium hydroxide hydrate (143.7 g) while keeping the temperature below 40 °C. The suspension was then stirred at ambient temperature overnight and filtered. To the obtained filtrate, concentrated hydrochloric acid (339 mL) was added while keeping the temperature below 30 °C. The suspension was aged for 2 h and filtered to give 4 as a white powder (95.6 g, NMR assay 100%; 52% yield).
Example 4:

Compound 2 (5.00 g) was dissolved in 2-propanol, dimethyl oxalate (7.02 g) was added and heated to 40 °C. Sodium methylate (25% in methanol; 15 mL) was slowly (10 min) added then the mixture was heated to 50-55 °C and stirred at that temperature for 72 h. The mixture was concentrated and components were separated by flash column chromatography (ethyl acetate/methanol 9:1 to 6:4). Early fractions gave compound 22 upon concentration, late fractions gave compound 23.
Compound 22: 1H NMR (DMSO-d6): δ 2.49 (m, 2H), 3.28 (s, 6H), 3.73 (s, 3H), 3.85 (s, 3H), 4.41 (m, 1 H), 4.50 (m, 1 H), 6.65 (s, 1 H), 8.36 (s, 1 H). 13C NMR (DMSO-d6): δ 51.63, 53.36, 54.25, 55.47, 102.71 , 1 18.24, 123.60, 140.81 , 150.21 , 162.44, 164.49, 173.43.
Compound 23: 1H NMR (DMSO-d6): δ 2.49 (m, 2H), 3.26 (s, 6H); 3.70 (s, 3H); 4.33 (d, 1 H); 4.60 (m, 1 H), 6.19 (s, 1 H), 8.12 (s, 1 H). 13C NMR (DMSO-d6): δ 50.03, 51.34, 54.59, 54.85, 102.91 , 1 16.04, 1 18.19, 148.32, 152.12, 163.46, 165.24, 174.99
Example 5:

Compound 3 (5.5 g; assay 53%) was suspended in acetonitrile, acetic acid (6 mL) and methanesulfonic acid (2.5 mL) were added followed by the heating of mixture to 70 °C for 4 h. The suspension was filtered and filtrate cooled to ambient temperature. Triethylamine (6.6 mL) and (R)-3-amino-butan-1 -ol (1.24 mL) was added followed by heating the mixture at reflux temperature for 20-24 h. The mixture was filtered, filtrate concentrated and 1 M HCI (100 mL) was added, followed by extraction with dichloromethane (3 x 50 mL). Combined organic fractions were concentrated, 2-propanol was added (10 mL) and suspension was stirred at 70-80 °C for 10 min, left to cool to ambient temperature then filtered to give 2.19 g of compound 4 (73%).
1H NMR (DMSO-de): δ 1.31 (d, 3H), 1.52 (m, 1 H), 1 .97 (m, 1 H), 3.89 (m, 1 H), 4.01 (m, 1 H), 4.46 (m, 1 H), 4.64 (m, 1 H), 4.78 (m, 1 H), 5.50 (m, 1 H), 7.29 (s, 1 H), 8.88 (s, 1 H), 15.83 (s, 1 H). 13C NMR (DMSO-d6): δ 15.22, 29.14, 45.26, 51.13, 62.09, 76.03, 1 16.31 , 1 18.79, 140.53, 146.79, 155.36, 165.24, 178.75.
Example 6:

Compound 3 (14.55 g; assay 49%) was suspended in acetonitrile (125 mL), acetic acid (15 mL) and methanesulfonic acid (6.25 mL) were added followed by the heating of mixture to 70 °C for 4 h. The suspension was filtered and filtrate cooled to ambient temperature. Triethylamine (16.5 mL) and (S)-2-aminopropanol (2.45 mL) was added followed by heating the mixture at reflux temperature for 24 h. The insoluble product was filtered, washed with 2-propanol (20 mL) and dried to give (3S, 1 1 aR)-3-methyl-5,7-dioxo-2,3,5,7, 1 1 ,1 1 a-hexahydrooxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxylic acid (5.2 g, 75%).
1H NMR (DMSO-d6): δ 1.31 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.8 Hz, 1 H), 4.13 (dd, J = 1 1.7, 10.3 Hz, 1 H), 4.28 (m, 1 H), 4.39 (dd, J = 8.6, 6.8 Hz, 1 H), 4.92 (dd, J = 12.3, 4.2 Hz, 1 H), 5.45 (dd, J = 10.2, 4.1 Hz, 1 H), 7.16 (s, 1 H), 8.84 (s, 1 H), 15.74 (s, 1 H).
Example 7:

Compound 4 (0.63 g) was dissolved in dichloromethane (15 mL), cooled to 5°C, then triethylamine (0.31 mL) was added, followed by ethyl chloroformate (0.26 mL), followed by slow (30 min) addition of 2,4-difluorobenzylamine. The mixture was then stirred at ambient temperature for 24 h. Water (10 mL) was added, organic phase was separated and washed with 1 M HCI (15 mL) and water (15 mL), concentrated and treated with 2-propanol to give the product 5 in a quantitative yield.
1H NMR (CDCI3): δ 1.39 (d, 3H), 1.52 (s, 1 H), 2.19 (m, 1 H), 4.00 (m, 2H), 4.16 (m, 1 H), 4.31 (m, 1 H), 4.62 (d, 2H), 5.00 (m, 1 H), 5.27 (m, 1 H), 6.80 (m 2H), 7.33 (m, 2H), 8.49 (s, 1 H), 10.48 (s, 1 H). 13C NMR (CDCI3): 15.50, 29.22, 36.43, 45.19, 51.83, 62.79, 103.71 , 103.91 , 1 1 1 .0, 1 1 1 .18, 120.59, 123.04, 130.40, 137.41 , 144.58, 156.27, 163. 87, 177.83.
Example 8:

To a suspension of 4 (2.84 g, 10 mmol) in a mixture of triethylamine (2.24 mL, 16 mmol) and acetone (50 mL) stirring on an ice bath was added ethyl chloroformate (1 .20 mL, 12 mmol). After stirring for 10 min, 2,4-difluorobenzylamine (1.21 mL, 10 mmol) was added and the mixture left stirring at room temperature for 1 h. The product was isolated by slowly diluting the reaction mixture with water (50 mL), partial concentration, filtration, washing with water (2 50 mL) and drying. There was obtained 5 as a white powder (3.48 g, 86%): mp 181.0-184.7 °C. 1H NMR (DMSO-d6): δ 1.29 (d, J = 7.0 Hz, 3H), 1 .56 (dd, J = 13.9, 2.0 Hz, 1 H), 1 .93-2.06 (m, 1 H), 3.90 (ddd, J = 1 1.6, 5.0, 2.1 Hz, 1 H), 3.98 (td, J = 12.0, 2.2 Hz, 1 H), 4.45 (dd, J = 13.6, 6.6 Hz, 1 H), 4.72 (dd, J = 13.6, 3.8 Hz, 1 H), 4.74-4.81 (m, 1 H), 5.44 (dd, J = 6.6, 3.8 Hz, 1 H), 8.93 (s, 1 H), 15.14 (s, 1 H). 13C NMR (DMSO-d6): δ 15.78, 29.13, 44.89, 52.88, 61 .63, 75.61 , 1 13.54, 128.49, 136.42, 145.64, 154.62, 164.58, 174.58
Example 9:

To a suspension of 4 (1 1.36 g, 40 mmol) in acetonitrile (80 mL) stirring at room temperature was added TCCA (9.29 g, 38 mmol) and DABCO (0.23 g, 5 mol%). After stirring at room temperature for 1 h, the reaction was quenched with a mixture of DMSO (5.26 mL) and water (1.33 mL). The insoluble cyanuric acid was removed by filtration and the filtrate evaporated under reduced pressure to give viscous oil. This was triturated in methanol (20 mL) to induce crystallization. The product was filtered, washed with cold methanol (10 mL) and dried to give 7 as a yellowish powder (5.13 g, 41 %): mp 191 .3-198.7 °C.
Example 10:

Attempted chlorination of 23: Compound 23 (0.54g) was suspended in acetonitrile (10 mL) and trichlorocyanuric acid (0.44 g) was added and the solution was stirred at ambient temperature overnight. Precipitate was filtered. Only traces of a product corresponding to the compound 26 could be detected in the reaction mixture by LC-MS analysis. Conversion did not improve with time.
Example 11 :

Attempted chlorination of 3: Compound 3 (0.30 g) was suspended in acetonitrile (5 mL) and trichlorocyanuric acid (0.13 g) was added. The suspension was stirred at ambient temperature overnight. Only traces of a product corresponding to the compound 24 could be detected in the reaction mixture by LC-MS analysis.
Example 12:

9 10
Trichloroisocyanuric acid (0.23 g) was added in a single portion to a stirred solution of the diethyl 1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (9, 0.66 g) in dry acetonitrile (4 mL) at room temperature. The resulting suspension was stirred at room temperature for ca. 24 h. The reaction mixture was diluted with dichloromethane and filtrated. The filtrate was then concentrated in vacuo to afford crude oil (0.86 g). Purification by flash chromatography (eluting ethyl acetate/cyclohexane) furnished diethyl 3-chloro-1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate, 10 as a yellow semi-solid (0.38 g). 1H NMR (CDCI3): δ 1.28 (t, J=7A Hz, 3H), 1 .37 (t, J=7.2 Hz, 3H), 3.35 (s, 6H), 3.89 (d, J=5.0 Hz, 2H), 4.27 (q, J=l A Hz, 2H), 4.43 (q, J=l A Hz, 2H), 4.48 (t, J=4.9 Hz, 1 H), 8.15 (s, 1 H). 13C NMR (CDCI3): δ 13.83, 14.13, 55.82, 57.09, 61.41 , 63.72, 102.52, 1 17.35, 126.90, 140.22, 146.92, 160.67, 164.13, 168.95.
Example 13:

Diethyl 1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (9, 0.64 g) was dissolved in anhydrous acetonitrile (6 mL) and treated sequentially with acetic acid (560 μί) and methanesulfonic acid (40 μί). The resulting mixture was heated to 62 °C and stirred for 4 h and more methanesulfonic acid (40 μΙ_) was added. After additional 2 h, more methanesulfonic acid (80 μΙ_) was added. This was repeated after additional 2 h, when more methanesulfonic acid (80 μΙ_) was added. The reaction mixture was stirred additional 17 h at 62 °C then was treated with a mixture of (R)-3-aminobutanol (0.22 g), triethylamine (0.5 mL) and acetonitrile (0.7 mL). The reaction mixture was stirred additional 22 h at 62 °C and then concentrated in vacuo. The crude material was partitioned between dichloromethane and 1 M HCI solution (15 mL). The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford the crude (4R, 12aS)-ethyl 4-methyl-6,8-dioxo-3,4,6,8, 12,12a-hexahydro-2H-pyrido[1 ',2':4,5]pyrazino[2, 1 -b][1 ,3]oxazine-9-carboxylate (11 ) as a brownish oil (0.61 g).
1H NMR (CD3OD): δ 8.44 (s, 1 H), 7.16 (m, 1 H), 5.48 (t, J=4.8 Hz, 1 H), 4.86 (m, 1 H), 4.49 (dd, J=13.6, 4.0 Hz, 1 H), 4.30-4.25 (m, 3H), 4.09 (dt, J=12.1 , 2.3 Hz, 1 H), 3.96 (ddd, J=1 1.7, 5.0, 2.1 Hz, 1 H), 2.18-2.10 (m, 1 H), 1.60-1 .56 (m, 1 H) 1 .39 (d, J=7A Hz, 3H), 1.33 (t, J=7A Hz, 3H). 13C NMR (CDCI3): δ 8.45, 14.08, 15.39, 29.17, 45.04, 45.72, 51 .56, 60.86, 62.61 , 76.33, 1 19.54, 123.72, 136.96, 145.67, 156.26, 163.68, 175.43
Example 14:

10
Diethyl 3-chloro-1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (10, 1.23 g) was dissolved in 85% formic acid (25 mL) at room temperature. The mixture was warmed to 40 °C and stirred for 23 h. The reaction mixture was concentrated in vacuo, and then partitioned between dichloromethane and aqueous NaHC03 solution. The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford brownish oil (0.49 g). The crude oil was dissolved in anhydrous toluene (5 mL) and treated sequentially with (R)-3-aminobutanol (0.19 g), methanol (0.2 mL) and acetic acid (96 μί). The resulting mixture was heated to 90 °C and stirred for 20 h. The reaction mixture was cooled to room temperature and then partitioned between dichloromethane and aqueous NaHC03 solution. The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford the crude (4R,12aS)-Ethyl 7-chloro-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1 ',2':4,5] pyrazino [2, 1-b][1 ,3]oxazine-9-carboxylate (12) as a brownish oil (0.24 g).
Example 15:

To a solution of 4 (5.68 g, 20 mmol) in dichloromethane (50 mL) stirring in an ice bath was added triethylamine (5.6 mL, 40 mmol), followed by ethyl chloroformate (2.61 mL, 26 mmol). After 20 min, ethanol (50 mL) was added. The mixture was then left stirring 24 h at room temperature and concentrated under reduced pressure. The residue was triturated in acetone (80 mL). The insoluble salt (triethylamine hydrochloride) was removed by filtration. The filtrate was evaporated under reduced pressure to give 11 as an amorphous solid in a quantitative yield (6.1 g).
Example 16:

To a stirring solution of 11 (0.94 g, 3.0 mmol) in acetonitrile (8 mL) heated at 40 °C was added TCCA in portions during 1 h (0.44 g, 1 .8 mmol). After an additional 1 h, the reaction mixture was diluted with a solution of NaHS03 (0.60 g) in water (60 mL), extracted with dichloromethane (50 mL) and the extract evaporated under reduced pressure to give a crude product which was purified by flash chromatography (CH2CI2 : MeOH, from 98 : 2 to 80 : 20) to give 12 (0.45 g, 44%).
1H NMR (CDCI3): δ 1.37 (t, J = 7.1 Hz, 3H), 1.38 (d, J = 7.0 Hz, 3H), 1 .56 (dq, J = 13.9, 2.2 Hz, 1 H), 2.21 (m, 1 H), 3.99 (d, J = 2.3 Hz, 1 H), 4.00 (t, J = 1.8 Hz, 1 H), 4.10 (dd, J = 13.2, 6.6 Hz, 1 H), 4.37-4.27 (m, 3H), 4.98 (m, 1 H), 5.35 (dd, J = 6.6, 3.8 Hz, 1 H), 8.07 (s, 1 H).
13C NMR (CDCI3): δ 14.20, 16.09, 29.34, 44.87, 53.73, 61.49, 62.29, 76.01 , 1 16.22, 133.1 1 , 134.18, 144.52, 155.48, 163.88, 169.98.
Example 17:

To a mixture of 7 (3.89 g, 12.2 mmol) in methanol (12 mL) was added sodium methylate (22.3 mL, 97.6 mmol). The reaction mixture was stirred for 24 h at 30 °C and then quenched with a slow addition of 3M hydrochloric acid (35 mL) while stirring in an ice bath. The mixture was concentrated under reduced pressure to remove most of the methanol, then extracted with dichloromethane (2 30 mL), the combined extracts washed with water (30 mL) and evaporated under reduced pressure. Methanol (20 mL) was added to the obtained amorphous residue and removed under reduced pressure to yield the solid 8 (3.69 g, 98%).
1H NMR (CDCI3): δ 15.04 (s, 1 H), 8.42 (s, 1 H), 5.29 (dd, J=5.6, 3.9 Hz, 1 H), 5.01 -4.96 (m, 1 H), 4.42 (dd, J=13.6, 3.6 Hz, 1 H), 4.25 (dd, J=13.6, 6.0 Hz, 1 H), 4.05 (s, 3H), 4.00-3.97 (m, 2H), 2.21 -2-14 (m, 1 H), 1.53 (dd, J=14.1 , 1.9 Hz, 1 H), 1.36 (d, J=7 Hz, 3H). 13C NMR (CDCI3): δ 176.35, 165.94, 155.03, 153.70, 143.08, 130.90, 1 15.94, 76.05, 62.65, 61.45, 53.86, 44.96, 29.43, 16.06.
Example 18:

To a suspension of 7 (2.55 g, 8.0 mmol) in a mixture of triethylamine (1 .46 mL, 10.4 mmol) and acetone (32 mL) stirring on an ice bath was added ethyl chloroformate (0.88 mL, 8.8 mmol). After stirring for 10 min, 2,4-difluorobenzylamine (1.07 mL, 8.8 mmol) was added and the mixture left stirring at room temperature for 1 h. The product was isolated by slowly diluting the reaction mixture with water (40 mL), filtration, washing with water (2 30 mL) and drying. There was obtained 2.91 g of 6 as a white powder (83%).
1H NMR (CDCI3): δ 1.30 (d, J = 7.0 Hz, 3H), 1 .49 (dd, J = 14.0, 2.2 Hz, 1 H), 2.14 (ddd, J = 14.6, 1 1.1 , 6.4 Hz, 1 H), 3.89-3.95 (m, 2H), 4.09-4.15 (m, 1 H), 4.26 (dd, J = 13.4, 3.8 Hz, 1 H), 4.55 (d, J = 5.8 Hz, 2H), 4.89-4.98 (m, 1 H), 5.18 (dd, J = 6.2, 3.8 Hz, 1 H), 6.68-6.79 (m, 2H), 7.23-7.31 (m, 1 H), 8.41 (s, 1 H), 10.24 (t, J = 5.8 Hz, 1 H). 13C NMR (CDCI3): δ 16.09, 26.95, 29.30, 36.79, 45.1 1 , 45.28, 53.86, 62.47, 75.93, 103.87 (t, J = 25.4 Hz), 1 1 1 .21 (dd, J = 21 .0, 3.4 Hz), 1 17.32, 130.58 (dd, J = 9.3, 5.8 Hz), 133.40, 143.54, 155.34, 163.16, 163.25, 163.35, 172.88.
Example 19:

To a suspension of 5 (1 .67 g, 4 mmol) in acetonitrile (20 mL) was added DABCO (23 mg, 5 mol%) and TCCA (0.62 g, 2.52 mmol). The mixture was stirred 18 h at 40 °C protected from light and then quenched with a mixture of DMSO (0.48 mL) and water (0.12 mL). The insoluble cyanuric acid was removed by filtration and washed with acetonitrile (5 mL). The filtrate was evaporated under reduced pressure to give viscous oil that was crystallized from a mixture of methanol (6 mL) and water (3 mL), by slowly cooling the solution from 60 °C to room
temperature. The product 6 was filtered, washed with cold methanol (5 mL) and dried to give an off-white powder (1.07 g, 61 %).
1H NMR (CDCI3): δ 1.30 (d, J = 7.0 Hz, 3H), 1 .49 (dd, J = 14.0, 2.2 Hz, 1 H), 2.14 (ddd, J = 14.6, 1 1.1 , 6.4 Hz, 1 H), 3.89-3.95 (m, 2H), 4.09-4.15 (m, 1 H), 4.26 (dd, J = 13.4, 3.8 Hz, 1 H), 4.55 (d, J = 5.8 Hz, 2H), 4.89-4.98 (m, 1 H), 5.18 (dd, J = 6.2, 3.8 Hz, 1 H), 6.68-6.79 (m, 2H), 7.23-7.31 (m, 1 H), 8.41 (s, 1 H), 10.24 (t, J = 5.8 Hz, 1 H). 13C NMR (CDCI3): δ 16.09, 26.95, 29.30, 36.79, 45.1 1 , 45.28, 53.86, 62.47, 75.93, 103.87 (t, J = 25.4 Hz), 1 1 1 .21 (dd, J = 21.0, 3.4 Hz), 1 17.32, 130.58 (dd, J = 9.3, 5.8 Hz), 133.40, 143.54, 155.34, 163.16, 163.25, 163.35, 172.88.
Example 20:

To a suspension of 6 (0.44 g) in anhydrous methanol (1 mL) was added a 25% methanolic solution of sodium methylate (1 .14 mL) and the mixture stirred for 4 h at 40 °C. The reaction was quenched with acetic acid (0.4 mL), diluted with water (8 mL), extracted with 2-methyltetrahydrofuran (12 mL), the extract washed with 1 M NaOH(aq) (8 mL), water (8 mL) and evaporated under reduced pressure. To the oily residue was added methanol (8 mL) and evaporated under reduced pressure to give 27 as a white solid (0.38 g, 88%).
Example 21 :

The suspension of (4R, 12aS)-7-chloro-N-(2,4-difluorobenzyl)-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1 ',2':4,5]pyrazino[2, 1 -b][1 ,3]oxazine-9-carboxamide (6, 0.44 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2 mL) was stirred at room temperature for 24 h. The reaction was quenched with 2M H2S04 (1 .18 mL) and left stirring for 2 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with water (2 x 2 mL). The obtained white precipitate (0.38 g) was suspended in THF-water (1 :1 , 4.5 mL) and stirred at room temperature for ca. 2 h. The reaction mixture was filtered through fitted funnel rinsing with water (2 χ 1 mL) and dried in vacuo at 40°C to afford pure dolutegravir as a white solid (0.33 g, HPLC purity: 99.38%).
1H NMR (DMSO-d6): δ 12.51 (s, 1 H), 10.36 (t, J=5.9 Hz, 1 H), 8.50 (s, 1 H), 7.41-7.36 (m, 1 H), 7.26-7.21 (m, 1 H), 7.07-7.03 (m, 1 H), 5.45 (dd, J=5.4, 4.3 Hz, 1 H), 4.81 -4.76 (m, 1 H), 4.59-4.53 (m, 3H), 4.36 (dd, J=13.8, 5.8 Hz, 1 H), 4.05-4.00 (m, 1 H), 3.91-3.88 (m, 1 H), 2.05-1 .97 (m, 1 H), 1.55-1.52 (m, 1 H), 1 .33 (d, J=7.1 Hz, 3H). 13C NMR (DMSO-d6): δ 170.27, 163.68, 162.29, 161 .78 (dd), 159.82 (dd), 154.61 , 140.64, 130.74 (d), 130.67 (d), 122.37 (d), 1 16.73, 1 15.38, 1 1 1 .33 (d), 103.80 (t), 62.01 , 51 .16, 44.69, 35.74, 29.13, 15.21.
Example 22:

A suspension of dolutegravir (0.31 g) in methanol (4 mL) was cooled to 0 °C.25% Solution of sodium methoxide in methanol was added to the mixture and the resulting suspension was stirred at 0 °C for 2 h, then at room temperature for 23 h. The reaction mixture was then filtered through fitted funnel rinsing with methanol (3 x 10 mL). The white precipitate was dried overnight at room temperature to afford pure dolutegravir sodium as a white solid (0.26 g, HPLC purity: 99.84%).
1H NMR (DMSO-d6): δ 10.70 (t, J=5.8, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J=6.4Hz, 1H), 4.51 (d, J=5.5Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J=14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J=12.9 Hz, 1H), 1.24 (d, J=7.0Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 23:

The suspension of 6 (0.44 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2 mL) was stirred at room temperature for 24 h. The reaction was diluted with absolute ethanol (10 mL) and left stirring for ca. 30 min at room temperature. The reaction mixture was filtered through fitted funnel rinsing with absolute ethanol (3 x 10 mL) and dried in vacuo at room temperature to afford dolutegravir sodium as a pale yellow solid (0.43 g, HPLC purity: 98.80%). 1H NMR (DMSO-d6): δ 10.70 (t, J = 5.8 Hz, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J = 6.4 Hz, 1H), 4.51 (d, J = 5.5 Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J= 14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J = 12.9 Hz, 1H), 1.24 (d, J = 7.0 Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 24:

The suspension of (4R,12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1',2':4,5]pyrazino[2,1-6][1,3]oxazine-9-carboxamide (27, 0.43 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2.5 mL) was stirred at room temperature for ca.24 h. The reaction was diluted with mixture of water/ethanol (5 mL, 1:1) and left stirring for ca. 1.5 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with mixture of water/ethanol (3 x 5 mL, 1:1) and dried in vacuo at room temperature to afford 15 as a pale yellow solid (0.41 g, HPLC purity: 98.87%).
1H NMR (DMSO-de): δ 10.70 (t, J = 5.8 Hz, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J = 6.4 Hz, 1H), 4.51 (d, J = 5.5 Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J = 14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J = 12.9 Hz, 1H), 1.24 (d, J = 7.0 Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 25:

The suspension of {4R, 12aS)-7-chloro-4-methyl-6,8-dioxo-3,4, 6,8, 12,12a-hexahydro-2H-pyrido[1',2':4,5]pyrazino[2,1-6][1,3]oxazine-9-carboxylic acid (7, 0.31 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2.5 mL) was stirred at 50 °C for 3 days. The reaction was quenched with 2M H2S04 (1.2 mL) and left stirring for 7 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with water (3x5 mL) and ethanol (5 mL) dried in vacuo at 40°C to afford 28 as a pale yellow solid (0.17 g).
1H NMR (DMSO-d6): δ 15.37 (s, 1H), 12.76 (s, 1H), 8.66 (s, 1H), 5.51-5.49 (m, 1H), 4.80-4.78 (m, 1H), 4.65 (dd, J=13.8, 3.7 Hz, 1H), 4.43 (dd, J=13.8, 5.9 Hz, 1H), 4.05 (t, J^^.b Hz, 1H), 3.91 (dd, J=11.4, 3.1 Hz, 1H), 2.07-2.00 (m, 1H), 1.56 (d, J=13.8 Hz, 1H), 1.34 (d, J=7.0 Hz, 3H).13C NMR (DMSO-de): δ 172.21, 165.39, 161.73, 153.61, 141.11, 118.66, 112.99, 75.95, 62.03, 51.50, 44.90, 29.08, 15.18.
Example 26:

The suspension of (4R,12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8, 12, 12a-hexahydro-2H-pyrido[1 ',2':4,5]pyrazino[2, 1 ,3]oxazine-9-carboxamide (27, 0.88 g) and solid sodium hydroxide (0.24 g) in absolute ethanol (20 mL) was stirred at 30 °C for 1.5 h. The reaction was quenched with 2M H2S04 (1 .5 mL) and left stirring for 3 hours at room temperature. The reaction mixture was filtered through fritted funnel and rinsed with water (3 x 2 mL) and ethanol (4 mL), and dried in vacuo at 40 °C to afford O-ethyl dolutegravir (29) as a pale yellow solid (0.25 g). The filtrate was extracted with ethyl acetate (3 x 5 mL). The combined organic layers were dried over MgS04, filtered and concentrated, then dried in vacuo at 40 °C to afford more 29 as a pale yellow solid (0.27 g).
1H NMR (CDCI3): δ 10.37 (t, J = 5.8 Hz, 1 H), 8.36 (s, 1 H), 7.37-7.32 (m, 1 H), 6.83-6.77 (m, 2H), 5.19 (dd, J = 5.9, 3.8 Hz, 1 H), 5.04-4.98 (m, 1 H), 4.61 (d, J = 6Hz, 2H), 4.26-4.22 (m, 3H), 4.1 1 (dd, J = 13.4, 5.9 Hz, 1 H), 3.97 (t, J = 2.4 Hz, 1 H), 3.96 (d, J = 2.4 Hz, 1 H), 2.21-2.14 (m, 1 H), 1.51 (dq, J = 14.0, 2.3 Hz, 1 H), 1 .47 (t, J = 7.0 Hz, 3H), 1 .35 (d, J = 7.1 Hz, 3H).
13C NMR (CDCI3): δ 174.78, 164.17, 162.49 (dd), 160.51 (dd), 155.72, 154.08, 142.32, 130.60 (dd), 129.33, 121 .51 (dd), 1 18.67, 1 1 1 .23 (dd), 103.78 (t), 76.15, 69.74, 62.58, 53.42, 44.58, 36.50 (d), 29.44, 16.04, 15.64.
Example 27:

The suspension of (4R, 12aS)-7-(benzyloxy)-4-methyl-3,4, 12,12a-tetrahydro-2H-pyrido[1 ',2':4,5]pyrazino[2, 1-b][1 ,3]oxazine-6,8-dione (30, 0.68 g, prepared according to prior art) and solid sodium hydroxide (0.40 g) in absolute ethanol (5 mL) was stirred at 50 °C for 14 h. The reaction was quenched with formic acid (0.35 mL), water (2 mL) was added and mixture was left stirring for additional 1 h at room temperature. The reaction mixture was extracted with ethyl acetate (3 x 5 mL) and the combined organic layers concentrated to afford a crude oil. Purification by flash chromatography (eluting with CH2CI2/methanol) afforded 32 as an orange solid (0.26 g, 52 %).
The above procedure if done at room temperature in same time period, affords 31 as orange oil (0.24 g, 43 %).
Compound 32: 1H NMR (DMSO-d6): δ 7.64 (d, J = 7.4 Hz, 1 H), 6.20 (d, J = 7.3 Hz, 1 H), 5.40 (dd, J = 5.1 , 4.2 Hz, 1 H), 4.83-4.78 (m, 1 H), 4.35 (dd, J = 13.6, 3.9 Hz, 1 H), 4.13 (dd, J = 13.6, 5.4 Hz, 1 H), 4.05-4.00 (m, 1 H), 3.90-3.85 (m, 1 H), 2.03-1.95 (m, 1 H), 1.52 (dd, J = 13.9, 1 .9 Hz, 1 H), 1.33 (d, J = 7.1 Hz, 3H). 13C NMR (DMSO-d6): δ 170.96, 163.01 , 153.48, 137.96, 1 16.83, 1 13.52, 76.18, 62.05, 50.39, 44.53, 29.21 , 15.28.
Compound 31 : 1H NMR (DMSO-d6): δ 7.67 (d, J = 7.4 Hz, 1 H), 6.28 (d, J = 7.4 Hz, 1 H), 5.29 (dd, J = 5.4, 3.8 Hz, 1 H), 4.82-4.75 (m, 1 H), 4.32 (dd, J = 13.6, 3.6 Hz, 1 H), 4.10 (dd, J = 13.5, 5.6 Hz, 1 H), 4.03-3.93 (m, 3H), 3.85 (ddd, J = 1 1 .6, 5.0, 2.2 Hz, 1 H), 1.97-1 .89 (m, 1 H), 1 .48 (dd, J = 13.8, 2.1 Hz, 1 H), 1.27 (d, J = 7.1 Hz, 3H), 1.26 (d, J = 7.0 Hz, 3H). 13C NMR (DMSO-d6): δ 174.38, 156.1 1 , 150.82, 139.48, 1 16.39, 1 13.52, 75.92, 67.31 , 61 .80, 51 .36, 44.22, 29.29, 15.76, 15.36.
Exa

The transformation of 6 to dolutegravir with sodium hydroxide in ethanol was monitored for the interconversion of intermediates. The suspension of 6 (0.44 g) and solid sodium hydroxide (0.20 g) in ethanol (3.33 ml.) was stirred at 22 °C. Samples of the reaction mixture were taken after 3, 8 and 24 h for UPLC analysis. After 24 h, the reaction mixture was quenched with 2 M H2S04 (5 ml_), and left stirring at room temperature. The reaction mixture was filtered through fritted funnel, the product rinsed with water (30 ml.) and dried in vacuo at 50 °C overnight to afford dolutegravir as a white solid (0.27 g, 64 %).
The results of reaction monitoring:
Time UPLC analysis (area%)
Entry
(h) compound 6 compound 29 dolutegravir
1 3 h 37.50 20.63 39.99
2 8 h 0.78 15.46 80.32
3 24h 0.31 8.56 88.21
Example 29:

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 27 (0.86 g) in MeOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2 ml.) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring demethylation of 27 in MeOH:

Example 30:

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 6 (0.88 g) in EtOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2 mL) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring of the transformations of 6 in ethanol with NaOH:

dol. = dolutegravir
Exa

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 27 (0.88 g) in EtOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2ml_) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring of the transformations of 27 in ethanol with NaOH:

dol. = dolutegravir
Example 32:

Compound 3 (30 g, 1 10 mmol; assay 99%) was suspended in acetonitrile (450 mL), acetic acid (73 mL) and methanesulfonic acid (25 mL) were added. The reaction mixture was stirred 4 h at 70 °C. The clear red solution was cooled to 25 °C. Triethylamine (77 mL) and (S)-2-aminopropanol (17 mL) were added and the mixture was stirred at reflux temperature for 20 h. The reaction mixture was cooled to 25 °C and the insoluble product filtered, washed with 1 M HCI(aq) (60 mL), water (3 * 60 mL) and dried to give 4c (19.49 g, 67%): mp = 313-315 °C; 1H NMR (DMSO-d6): δ 1.31 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.8 Hz, 1 H), 4.13 (dd, J = 1 1.7, 10.3 Hz, 1 H), 4.28 (m, 1 H), 4.39 (dd, J = 8.6, 6.8 Hz, 1 H), 4.92 (dd, J = 12.3, 4.2 Hz, 1 H), 5.45 (dd, J = 10.2, 4.1 Hz, 1 H), 7.16 (s, 1 H), 8.84 (s, 1 H), 15.74 (s, 1 H); 13C NMR (DMSO-d6) 16.5, 51.6, 52.9, 72.4, 81.6, 1 15.8, 1 18.1 , 141.5, 147.6, 153.4, 165.3, 179.0.
Example 33

Compound 4c (2.78 g) was suspended in dimethylformamide (40 mL), cooled to 0 °C, then triethylamine (3.52 mL) was added, followed by ethyl chloroformate (1 .31 mL). After 10 min there was added 2,4-difluorobenzylamine (1 .57 mL). The mixture was then stirred at 25 °C for 1 h. Water (150 mL) was added and the mixture extracted with dichloromethane (50 mL). The organic phase was separated, washed with water (2 χ 50 mL), dried over sodium sulfate and evaporated under reduced pressure. The residue (4.31 g) was treated with boiling 2-propanol (40 mL), the suspension cooled, the product filtered and dried to give the product 5c as a white powder (2.70 g, 69%): 99.80 area% by HPLC at 258 nm; mp = 222-223 °C; MS (ESI) m/z = 390 [MH]+; 1H NMR (DMSO-d6): δ 1 .30 (d, J = 6.3 Hz, 3H), 3.63 (dd, J = 8.6, 6.8 Hz, 1 H), 4.02 (m, 1 H), 4.26 (m, 1 H), 4.37 (dd, J = 8.6, 6.8 Hz, 1 H), 4.53 (d, J = 6.0 Hz, 2H), 4.84 (dd, J = 12.2, 4.2 Hz, 1 H), 5.40 (dd, J = 12.2, 4.2 Hz, 1 H), 6.91 (s, 1 H), 7.05 (m, 1 H), 7.24 (m, 1 H), 7.38 (m, 1 H), 8.62 (s, 1 H), 10.43 (t, J = 6.0 Hz, 1 H).

To a suspension of 5c (2.70 g, 6.9 mmol) in acetonitrile (32 mL) was added DABCO (39 mg, 5 mol%) and TCCA (1.01 g, 4.3 mmol). The mixture was stirred 20 h at 40 °C protected from light and then quenched with a mixture of DMSO (0.81 mL) and water (0.20 mL). The insoluble cyanuric acid was removed by filtration and washed with acetonitrile (10 mL). The filtrate was evaporated under reduced pressure to give viscous oil that was crystallized from a mixture of methanol (10 mL) and water (5 mL), by slowly cooling the solution from 60 °C to room temperature. The product 6c was filtered, washed with cold methanol (8 mL) and dried to give an off-white powder (1 .20 g, 41 %): mp = 225-227 °C; MS (ESI) m/z = 424 [MH]+; 1H NMR
(DMSO-d6): δ 1.28 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.9 Hz, 1 H), 4.09 (m, 1 H), 4.26 (m, 1 H), 4.35 (dd, J = 8.6, 6.6 Hz, 1 H), 4.54 (d, J = 5.9 Hz, 2H), 4.85 (dd, J = 12.3, 3.8 Hz, 1 H), 5.42 (dd, J = 10.1 , 3.8 Hz, 1 H), 7.06 (m, 1 H), 7.24 (m, 1 H), 7.40 (m, 1 H), 8.67 (s, 1 H), 10.24 (t, J = 6.0 Hz, 1 H).
Example 35

cabotegravir
The suspension of 6c (1.00 g, 2.4 mmol) and sodium hydroxide (0.57 g, 14.2 mmol) in absolute ethanol (7 mL) was stirred at 40 °C for 16 h. The reaction was quenched with 0.5M H2S04 (15 mL), extracted with dichloromethane (20 mL), the extract washed with water (20 mL) and evaporated under reduced pressure. The residue was triturated in MTBE (10 mL), the product filtered, washed with MTBE (10 mL) and dried to give cabotegravir as an off-white solid (0.74 g, 77%): MS (ESI) m/z = 405 [MH]+.
Lek, a Sandoz company, opens the first production facility in Slovenia for drug substances for innovative medicines at its Mengeš site

Dr Miro Cerar, the Prime Minister of the Republic of Slovenia
Lek, a Sandoz company, awarded for cooperation in practical training of students of the Faculty of Chemistry and Chemical Technology
30. 1. 2015
At a ceremony held on 22 January 2015 at the Faculty of Chemistry and Chemical Technology, University of Ljubljana, the Maks Samec awards and recognitions for 2014 were presented for the best doctoral thesis in the field of chemistry, the best doctoral thesis in the field of chemical engineering and chemical technology and for services and merits to the Faculty in the year 2014. On this occasion, the Faculty also wanted to thank all the companies and individuals who shared their knowledge and resources to help the Faculty on its education and research path.
Lek, a Sandoz company, received a plaque for taking part in the implementation of practical training, which was collected, on behalf of the company, by Samo Roš, Head of Human Resources and a Member of the Lek Board of Management. By doing so, the Faculty of Chemistry and Chemical Technology thanked all the mentors who directly transfer their expertise and valuable experience onto students, teaching them specific skills, encouraging their development, guiding them through the work process and ensuring that students become socialized in the workplace.
* * *
Lek, a Sandoz company, is one of key pillars of the second-largest generic pharmaceutical company globally. Its role within Sandoz is to act as: a leading global development center for technologically demanding products and technologies; a global manufacturing center for active pharmaceutical ingredients and medicines; a competence center for the development of vertically integrated products; a Sandoz competence center in the field of development and manufacturing of biosimilar products; and, a supply center for the markets of Central and Eastern Europe (CEE), South East Europe (SEE) and Commonwealth of Independent States (CIS), and it is responsible for sales on the Slovenian market. For further information please visit http://www.lek.si/en.Sandoz, the generic pharmaceuticals division of Novartis, is a global leader in the generic pharmaceutical sector. Sandoz employs over 26,400 employees and its products are available in more than 160 countries, offering a broad range of high-quality, affordable products that are no longer protected by patents. With USD 9.6 billion in sales in 2014, Sandoz has a portfolio of approximately 1,100 molecules, and holds the #1 position globally in biosimilars as well as in generic injectables, ophthalmics, dermatology and antibiotics, complemented by leading positions in the cardiovascular, metabolism, central nervous system, pain, gastrointestinal, respiratory, and hormonal therapeutic areas. Sandoz develops, produces, and markets these medicines, as well as active pharmaceutical and biotechnological substances. Nearly half of Sandoz’s portfolio is in differentiated products, which are defined as products that are more difficult to scientifically develop and manufacture than standard generics. In addition to strong organic growth since consolidating its generics businesses under the Sandoz brand name in 2003, Sandoz has benefitted from strong growth of its acquisitions, which include Lek (Slovenia), Sabex (Canada), Hexal (Germany), Eon Labs (US), EBEWE Pharma (Austria), Oriel Therapeutics (US), and Fougera Pharmaceuticals (US).
Sandoz is on Twitter. Sign up to follow @Sandoz_global at http://twitter.com/Sandoz_Global.
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, eye care, cost-saving generic pharmaceuticals, preventive vaccines and over-the-counter products. Novartis is the only global company with leading positions in these areas. In 2014, the Group achieved net sales of USD 58.0 billion, while R&D throughout the Group amounted to approximately USD 9.9 billion (USD 9.6 billion excluding impairment and amortization charges). Novartis Group companies employ approximately 130,000 full-time-equivalent associates. Novartis products are available in more than 180 countries around the world. For more information, please visit www.novartis.com
////////////Carbotegravir, Dolutegravir, New Patent, WO 2016113372, Lek Pharmaceutical and Chemical Co DD



























{kind=link}