
WO-2016147120, AZILSARTAN, NEW PATENT, SMILAX Laboratories Ltd
(WO2016147120) AN IMPROVED PROCESS FOR THE PREPARATION OF SUBSTANTIALLY PURE AZILSARTAN
Azilsartan (I) is an angiotensin receptor II antagonist used in the treatment of hypertension. Angiotensin II causes vasoconstriction via an angiotensin II receptor on the cell membrane and elevates blood pressure.
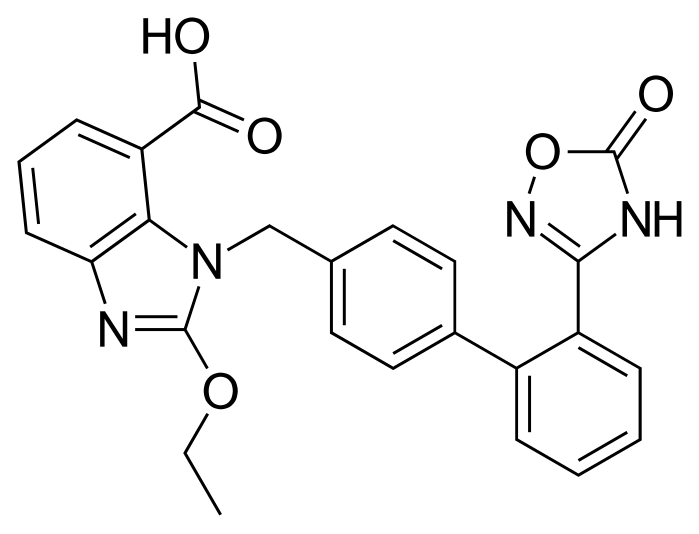
Azilsartan medoxomil i.e. (5-methyl-2-oxo-l,3-dioxol-4-yl)methyl 2-ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid is developed by Takeda pharmaceuticals and is marketed under the trade name Edarbi. It was approved by USFDA on 25 Feb, 2011 and EMEA on 7 Dec 2011 for the treatment of high blood pressure in adults.
Azilsartan medoxomil and its salts thereof are imbibed with properties such as strong and long lasting angiotensin II antagonistic activity and hypotensive action which has an insulin sensitizing activity useful for the treatment of metabolic diseases such as diabetes and the like., and a useful agent for the prophylaxis or treatment of circulatory diseases such as hypertension, cardiac diseases, nephritis and stroke. Azilsartan medoxomil is the prodrug of 2-Ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan).
Methods of preparing benzimidazole derivative useful as an angiotensin II receptor antagonist such as Azilsartan Medoxomil and salts thereof are disclosed by Takeda in US 5,243,054 (herein after referred as US '054 patent). The US'054 patent describes several synthetic routes for preparing Azilsartan. According to one of the synthetic process, the compound of formula II is reacted with hydroxylamine hydrochloride in a conventional organic solvent and sodium methoxide in methanol to give the amidoxime compound of formula III which on further reaction with ethyl chloroformate in presence of triethylamine base in refluxing xylene undergoes cyclization to provide a compound of formula IV. Azilsartan was prepared by hydrolysis of compound of formula IV in presence of lithium hydroxide by adjusting the pH with HC1. The process is as depicted below in Scheme A:


However, the amidoxime compound of formula III obtained by the above process contains about 50% of amide imputiy along with desired product, owing to the strong reaction conditions which impairs the quality and loss of yield. The pH adjustment with HC1 in the hydrolysis step of compound IV results in the formation of an undesired desethyl impurity of formula V due to acid sensitive nature of the ether linkage in the benzimidazole moiety of Azilsartan.
Formula V

According to another method disclosed in US'054 for the preparation of Azilsartan comprises by reacting ethoxycarboimidoyl biphenyl benzimidazole derivative of compound with ethyl chloroformate to give N-methoxycarbonyl ethoxycarboimidoyl biphenyl benzimidazole derivative, which is further converted to compound of formula IV and then to Azilsartan of formula I by hydrolysis.
According to one another embodiment method for the preparation of Azilsartan disclosed in US '054, cyanobiphenyl aminobenzoate derivative compound reacts with hydroxylamine hydrochloride in presence of triethylamine subsequently followed by addition of ethyl chlorocarbonate results in the formation of compound of formula IV which is further hydrolyzed to obtain Azilsartan of formula I.
J. Med. Chem. Vol. 39, No. 26, 5230-5237 (1996) describes the use of triethylamine as base during the conversion of compound of formula II to amidoxime compound of formula III and use of 2-ethylhexylchloroformate instead of ethylchloroformate as cyclizing agent.
Processes for the preparation of Azilsartan medoxomil and its potassium salt are described in US 7,157,584 which comprises reacting Azilsartan with 4-hydroxymethyl-5-methyl-l,3-dioxol-2-one in presence of dimethylacetamide, p-toluoyl sulfonylchloride, 4-dimethylaminopyridine and potassium carbonate.
PCT publication WO 2012/107814 discloses process for the preparation of Azilsartan or its esters or salts by reacting amidoxime compound of formula III with carbonyl source such as carbodiimides, dialkyl carbonate and phosgene equivalents in presence of a suitable base to obtain compound of formula IV which is further converted to Azilsartan and its pharmaceutically acceptable salts. The process for the preparation of Azilsartan is as depicted in Scheme B:

Scheme - B
This publication also discloses that use of a carbonyl source reduces the formation of the content of desethyl impurity during cyclization.
Polymorphs of Azilsartan and its salts are disclosed in WO 2013/044816 and WO 2013/186792.
All the above prior art methods for the preparation of Azilsartan have inherent disadvantages such as the usage of unsafe reagents, high boiling solvents, extreme reaction conditions invariably resulting in the formation of low pure intermediates as well as Azilsartan having a considerably higher content of desethyl impurity. Accordingly, there remains a need for the industrial preparation of substantially pure Azilsartan which is free of impurities with high yield.

Examples
Example-1: Preparation of Methyl-2-ethoxy-l-[[2'- ((hydroxycarbamimidoyl)biphenyl)-4-yl]methyl]-lH-benzimidazole-7-carboxylate (Formula-Ill):
To a stirred solution of DMSO (1500.0 mL), Hydroxylamine hydrochloride ( 126.7g 1.83mol) and Dipotassium hydrogen phosphate (634.9g 3.65mol) was added Methyl l-[[2'-cyanobiphenyl-4-yl]methyl]-2-ethoxybenzimidazole-7-carboxylate (lOO.Og 0.243mol) at 25-30°C. The reaction mass temperature was raised to 80-85°C and maintained for 30-40 hours. Reaction completion was monitored by TLC. Upon completion of reaction, reaction mass was cooled to 10- 15°C, and was poured into water (3000.0 mL), stirred for 45min at 20-25°C. and was filtered. The filtered wet solid was washed with water and dried at 65°C to get crude Methyl-2-ethoxy-l-[[(2'-(hydroxycarbarmrmdoyl)biphenyl-4-yl]methyl]-lH-benzimidazole-7-carboxylate. The wet material was slurried in Acetone (optional) at reflux and filtered at room temperature to obtain pure compound.
Yield: 79.92 g, 74.0%; HPLC Purity: 97.78%; Desethyl impurity: 0.318%; Amide impurity: 1.42%.
Example-2: Preparation of Methyl 2-ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (Formula-IV) :
To the pre cooled solution of Methylene dichloride (375.0 mL) and Methyl-2-ethoxy-1 -[[2' -((hydroxycarbamimidoyl)biphenyl)-4-yl] methyl]- lH-benzimidazole-7-carboxylate (75.0g, 0.168mol) was added ethyl chloroformate ( 18.3g 0.168mol)
followed by addition of triethylamine (18.75g 0.185mol). The reaction mass was maintained at 0-5 °C for about 1 hour. Upon completion of the reaction, reaction mass was poured into water (200.0 mL), organic layer was separated and washed with 5% NaHC03 solution (150.0 mL) and then with water (150.0 mL). The organic layer was dried over sodium sulfate and distilled to obtain the crude material (optionally be isolated using cyclohexane solvent). To this obtained crude material, ethyl acetate (750.0mL) and potassium carbonate (112.5g 0.814mol) were added and heated to reflux for 6 to 8 hours. The contents were cooled, filtered and wet solid was slurried in water. Wet material so obtained was slurried in ethyl acetate at reflux and filtered at room temperature and dried at 60-65°C to give Methyl 2-ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate.
Yield: 64.27 g, 81.0 %; HPLC Purity: 99.80%; Desethyl impurity: 0.085%.
Example-3: Preparation of 2-Ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan)
A mixture of 0.4N NaOH solution (395.8 mL) and Methyl 2-ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (25. Og) were stirred at 50-55°C for period of 60min. The reaction mass was cooled to room temperature and the product layer was washed with ethyl acetate (125.0mL). pH of the separated aqueous product layer was adjusted to 4.0 to 5.0 using dilute acetic acid at 0-5 °C. The obtained solid material was filtered and washed with water (lOO.OmL). This material was dried to obtain the title product.
Yield: 20.0 g, 82.47%; HPLC Purity : 99.80%; Desethyl impurity: 0.10%.
Example-4: Preparation of 2-Ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan)
A mixture of 0.4N NaOH solution (633.33 mL) and Methyl 2-ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (40.0g) were stirred at 50-55°C for period of 60min. The reaction mass was cooled to room temperature and the product layer was washed with ethyl acetate (200.0mL). pH of the separated aqueous product layer was adjusted to 4.0 to 4.5 using acetic acid at 10-15°C. The obtained solid material was filtered and washed with water (lOO.OmL). This material was dried to obtain the title product.
Yield: 32.35 g, 83.37%; HPLC Purity: 99.45%; Desethyl impurity: 0.12%.
Example-5: Preparation of 2-Ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan)
A mixture of 0.4N NaOH solution (791.66 mL) and Methyl 2-ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (50.0g) were stirred at 50-55°C for period of 60min. The reaction mass was cooled to room temperature and the product layer was washed with ethyl acetate (250.0mL). pH of the separated aqueous product layer was adjusted to 3.0 to 4.0 using citric acid at 10-15°C. The obtained solid material was filtered and washed with water (125.0mL). This material was dried to obtain the title product.
Yield: 37.0 g, 76.28%; HPLC Purity: 99.69%; Desethyl impurity: 0.083%.
Example-6: Preparation of 2-Ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan)
A mixture of 0.4N NaOH solution (395.83 mL) and Methyl 2-ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (25. Og) were stirred at 50-55°C for period of 60min. The reaction mass was cooled to room temperature and the product layer was washed with ethyl acetate (lOO.OmL). pH of the separated aqueous product layer was adjusted to 3.0 to 4.0
using hydrochloric acid at 10-15°C. The obtained solid material was filtered and washed with water (72.5 mL). This material was dried to obtain the title product. Yield: 20.22 g, 83.37%; HPLC Purity: 99.45%; Desethyl impurity: 0.217%.
Example-7: Purification of 2-Ethoxy-l-[[2'-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan)
Charged 2-Ethoxy- 1 - [[2' -(2,5-dihydro-5-oxo- 1 ,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (lOO.Og), methanol (600.0ml) and methylene dichloride (600.0ml) and were stirred for 10 min at 25-30°C to get a clear solution. Above solution was treated with Activated carbon (lO.Og) and stirred for 10.0 min at 25-30°C. Reaction mixture was passed through a hyflow bed and washed with a mixture of (1: 1) ratio of 200.0ml methanol and methylene dichloride. The solvent mixture was distilled out at below 50°C till the solid formation was observed. Reaction mixture is stirred for 30.0min at 30°C, then the solid was filtered and washed with 200.0ml of methylene dichloride. To the obtained solid, methanol (450.0 ml) was charged at 25-30°C, heated to 45°C, stirred for 30 min at 45°C and then cooled to 30°C. After cooling, the solid was filtered and washed with methanol (90.0ml) which was further dried at 50-55°C for 12 hours.
Yield: 80.0 g, 80.0%; HPLC Purity: 99.96%; Desethyl impurity : 0.012%.


Smilax Managing Director, S. Murali Krishna received the award from Hon’ble Chief Minister of Andhra Pradesh Shri. N. Kiran Kumar Reddy.
//////////WO 2016147120, AZILSARTAN, NEW PATENT, SMILAX Laboratories Ltd














