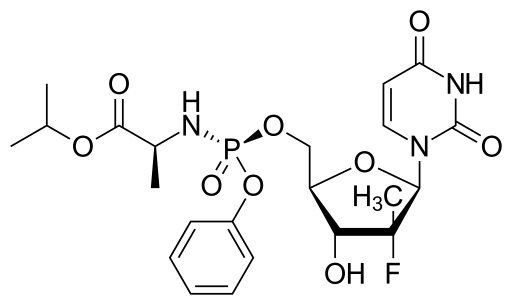
SOFOSBUVIR
NEW PATENT WO2015188782,
(WO2015188782) METHOD FOR PREPARING SOFOSBUVIR
CHIA TAI TIANQING PHARMACEUTICAL GROUP CO., LTD [CN/CN]; No. 8 Julong North Rd., Xinpu District Lianyungang, Jiangsu 222006 (CN)

Sofosbuvir synthesis routes currently used include the following two methods:
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015188782&redirectedID=true

Preparation Example 1 sofosbuvir implementation
[0067]
[0068]
Step (a):
[0069]
At 0 ℃, dichloro-phenyl phosphate (6.0g, 28.4mmol) in dry dichloromethane (30ml) and stirred added alanine isopropyl ester hydrochloride (4.8g, 28.4mmol), the mixture After stirring and cooling to -55 ℃, was slowly added dropwise triethylamine (6.5g, 64mmol) and dichloromethane (30ml) mixed solution, keeping the temperature during at -55 ℃, dropping was completed, stirring was continued for 60 minutes, after liters to -5 ℃ stirred for 2 hours, TLC monitored the reaction was complete. To remove triethylamine hydrochloride was filtered and the filtrate evaporated under reduced pressure to give compound 3-1 as a colorless oil (Sp / Rp = 1/1).
[0070]
31 PNMR (CDCl 3 , 300 Hz, H 3 PO 4 as internal standard): δ8.25 & 7.94 (1: 1);
[0071]
1 HNMR (CDCl 3 , 300 MHz): δ7.39-7.34 (m, 2H), 7.27-7.18 (m, 3H), 5.10-5.02 (m, 1H), 4.51 (br, 1H), 4.11 (m, 1H ), 1.49 (d, 3H), 1.29-1.24 (m, 6H);
[0072]
13 C NMR (CDCl 3 , 300 MHz): δ172.1 (Rp), 196.3 (Sp), 129.8,129.6 (d), 125.9,120.5 (d), 69.7 (d), 50.7 (d), 21.6 (d), 20.4 (d).
[0073]
Step (b):
[0074]
At 5 ℃, the compound of formula 2 (5.20g, 20.0mmol) in dry THF (30ml) and stirred at t-butyl chloride (1.0M THF solution, 42ml, 42.0mmol). The reaction temperature was raised to 25 ℃, and the mixture was stirred for 30 minutes. After addition of lithium chloride (21.0mmol), was slowly added dropwise the compound 3-1 (approximately 28.4mmol) and THF (30ml) mixed solution, keeping the temperature during at 5 ℃. Bi drops, stirred for 15 hours. With aqueous 1N HCl (25ml) The reaction solution was quenched (HPLC assay Sp: Rp ratio of 4: 1). Toluene was added (100ml), temperature was raised to room temperature. The organic layer was washed with 1N HCl, water, 5% Na 2 CO 3 and washed with brine, dried over anhydrous magnesium sulfate, filtered, and the solvent was distilled off under reduced pressure to a solid, was added methylene chloride (20ml), stirred for 5 minutes plus isopropyl ether, stirring was continued for 2 hours, the precipitated solid was filtered off. The solid was dissolved by heating in dichloromethane (60ml), slowly cooled to room temperature and the precipitated crystalline solid. Repeat if necessary obtain pure crystalline sofosbuvir (2.6g, yield 25%, HPLC purity measured 98.8%).
[0075]
31 PNMR (CDCl 3 , 300 Hz, H 3 PO 4 as internal standard): δ3.54ppm;
[0076]
13 C NMR (CDCl 3 , 300 Hz): δ173.1 (d), 162.7 (s), 150.2 (d), 139.3 (d), 129.6 (q);
[0077]
MS (M + H): 530.1.
[0078]
Preparation of compounds of formula 2 shown in Example 3-2
[0079]
[0080]
(1) a nucleophilic reagent as NaSCN, the phase transfer catalyst is TBAB
[0081]
The compound (product of Example 1, step (a)) is represented by the formula 3-1 is dissolved in dichloromethane (20ml) was added TBAB (2.8mmol), the NaSCN (35mmol) in water (2.0ml) was added dropwise It was added to the reaction solution. Dropping was completed, stirring was continued for 60 minutes, the solid was removed by filtration. The filtrate was washed with water, add MgSO 4 dried for 24 hours. Filtered, and the filtrate was evaporated under reduced pressure, to obtain a compound of formula 3-2 as (where X = SCN).
[0082]
1 HNMR (CDCl 3 , 500Hz): δ7.32-7.13 (m, 3H), 7.08-7.02 (m, 2H), 5.0-4.9 (m, 1H), 3.92 (m, 1H), 1.49 (m, 3H ), 1.23-1.17 (m, 6H);
[0083]
31 PNMR (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ-18.16 / -18.26.
[0084]
(2) nucleophile NaSCN, phase transfer catalyst is 18-crown-6 ether
[0085]
The compound (product of Example 1, step (a)) is represented by the formula 3-1 is dissolved in ethyl acetate (20ml) was added 18-crown -6 (2.8mmol), the NaSCN (35mmol) was added to the above the reaction mixture. Dropping was completed, stirring was continued for 60 minutes, the solid was removed by filtration. The filtrate was washed with water, add MgSO 4 dried for 24 hours. Filtered, and the filtrate was evaporated under reduced pressure, to obtain a compound of formula 3-2 as (where X = SCN).
[0086]
(3) nucleophile NaSCN, phase transfer catalyst is TBAB and 18-crown-6
[0087]
The compound (product of Example 1, step (a)) is represented by the formula 3-1 is dissolved in dichloromethane (20ml) was added TBAB (2.8mmol) and 18-crown -6 (2.8mmol), the NaSCN (35mmol) in water (2.0ml) was added to the reaction solution. Dropping was completed, stirring was continued for 60 minutes, the solid was removed by filtration. The filtrate was washed with water, add MgSO 4 dried for 24 hours. Filtered, and the filtrate was evaporated under reduced pressure, to obtain a compound of formula 3-2 as (where X = SCN).
[0088]
(4) nucleophile as NaN 3 , phase transfer catalyst is TBAB
[0089]
The compound (product of Example 1, step (a)) is represented by the formula 3-1 is dissolved in dichloromethane (20ml) was added TBAB (2.8mmol), the NaN 3 (35 mmol) in water (2.0ml) solution of was added dropwise to the reaction solution. Dropping was completed, stirring was continued for 60 minutes, the solid was removed by filtration. The filtrate was washed with water, add MgSO 4 dried for 24 hours. Filtered, and the filtrate was evaporated under reduced pressure, to obtain a compound of formula 3-2 as (where X = N 3 ).
[0090]
1 HNMR (CDCl 3 , 500Hz): δ7.30-7.33 (m, 2H), 7.27-7.21 (m, 3H), 5.10-5.05 (m, 1H), 4.12-4.00 (m, 1H), 1.43 (d , 3H), 1.28-1.17 (m, 6H);
[0091]
31 PNMR- (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ2.04 / 2.19.
[0092]
(5) the nucleophilic reagent is KCN, the phase transfer catalyst is TBAB
[0093]
The compound was dissolved in methylene chloride as in formula 3-1 (20ml), was added TBAB (2.8mmol), the KCN (35mmol) in water (2.0ml) was added dropwise to the reaction solution. Dropping was completed, stirring was continued for 60 minutes, the solid was removed by filtration. The filtrate was washed with water, add MgSO 4 dried for 24 hours. Filtered, and the filtrate was evaporated under reduced pressure to remove the solvent to give a compound as shown in Formula 3-2 (where X = CN).
[0094]
1 HNMR (CDCl 3 , 300 Hz): δ7.22-7.13 (m, 3H), 7.09-7.02 (m, 2H), 5.01-4.95 (m, 1H), 4.08-3.93 (m, 1H), 1.43-1.35 (m, 3H), 1.20-1.17 (m, 6H);
[0095]
31 PNMR (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ-2.71 / -2.93.
[0096]
Preparation Example 3 sofosbuvir implementation
[0097]
[0098]
(1) X is SCN
[0099]
Under 5 ℃, the compound (5.20g, 20.0mmol) as shown in Equation 2 in dry THF (30ml) in. T-butyl chloride was added with stirring (1.0M THF solution, 42ml, 42.0mmol). The reaction temperature was raised to 25 ℃, and the mixture was stirred for 30 minutes. After addition of lithium chloride (21.0mmol), was slowly added dropwise a compound of formula 3-2 (Preparation Example 2 28.4 mmol, obtained) and THF (30ml) mixed solution, keeping the temperature during at 5 ℃. After dropping was completed, the mixture was stirred for 15 hours. With aqueous 1N HCl (25ml) The reaction solution was quenched (HPLC assay Sp: Rp ratio of 6: 1). After further addition of toluene (100ml), temperature was raised to room temperature. The organic layer was washed with 1N HCl, water, 5% Na 2 CO 3 and washed with brine, dried over anhydrous magnesium sulfate, filtered, and the solvent was distilled off under reduced pressure to a solid, was added methylene chloride (20ml), stirred for 5 minutes plus isopropyl ether, stirring was continued for 2 hours, the precipitated solid was filtered off. The solid was dissolved by heating in dichloromethane (60ml), slowly cooled to room temperature and the precipitated crystalline solid. Repeat if necessary obtain pure crystalline sofosbuvir (3.6g, yield 34%, HPLC purity measured 98.7%).
[0100]
1 HNMR (CDCl 3 , 300 MHz): [delta] 8.63 (s, 1H, NH), 7.46 (d, 1H, C6-H), 7.36 (t, 2H, O-aromatic), 7.18-7.24 (m, 3H, m, P-aromatic), 6.20-6.14 (d, 1H, Cl'-H), 5.70-5.68 (d, 1H, C5-H), 5.05-4.97 (m, 1H, CH- (CH 3 ) 2 ) , 4.57-4.41 (m, 2H, C5'-H2), 4.12-4.09 (d, 1H, C3'-H), 4.06-3.79 (m, 3H, C3'-OH, C4'-H, Ala-CH -CH 3 ), 3.79 (s, 1H, Ala-NH), 1.44 (d, 3H, C2'-H3), 1.36-1.34 (d, 3H, Ala-CH 3 ), 1.25-1.23 (t, 6H, CH- (CH 3 ) 2 );
[0101]
P 31 NMR (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ3.56.
[0102]
(2) X is N 3
[0103]
Under 5 ℃, the compound (5.20g, 20.0mmol) as shown in Equation 2 in dry THF (30ml) in. T-butyl chloride was added with stirring (1.0M THF solution, 42ml, 42.0mmol). The reaction temperature was raised to 25 ℃, and the mixture was stirred for 30 minutes. Was added lithium chloride (21.0mmol), was slowly added dropwise after the compound of formula 3-2 obtained in Preparation Example 2 (about 28.4 mmol) and THF (30ml) mixed solution, keeping the temperature during at 5 ℃. Bi drops, stirred for 15 hours. With aqueous 1N HCl (25ml) The reaction solution was quenched (HPLC assay Sp: Rp ratio of 7: 1). After further addition of toluene (100ml), temperature was raised to room temperature. The organic layer was washed with 1N HCl, water, 5% Na 2 CO 3 and washed with brine, dried over anhydrous magnesium sulfate, filtered, and the solvent was distilled off under reduced pressure to a solid, was added methylene chloride (20ml), stirred for 5 minutes plus isopropyl ether, stirring was continued for 2 hours, the precipitated solid was filtered off. The solid was dissolved by heating in dichloromethane (60ml), slowly cooled to room temperature and the precipitated crystalline solid. Repeat if necessary obtain pure crystalline sofosbuvir (4.2g, yield 40%, HPLC purity measured 98.8%).
[0104]
1 HNMR (CDCl 3 , 300 MHz): [delta] 8.63 (s, 1H, NH), 7.46 (d, 1H, C6-H), 7.36 (t, 2H, O-aromatic), 7.18-7.24 (m, 3H, m, P-aromatic), 6.20-6.14 (d, 1H, Cl'-H), 5.70-5.68 (d, 1H, C5-H), 5.05-4.97 (m, 1H, CH- (CH 3 ) 2 ) , 4.57-4.41 (m, 2H, C5'-H2), 4.12-4.09 (d, 1H, C3'-H), 4.06-3.79 (m, 3H, C3'-OH, C4'-H, Ala-CH -CH 3 ), 3.79 (s, 1H, Ala-NH), 1.44 (d, 3H, C2'-H3), 1.36-1.34 (d, 3H, Ala-CH 3 ), 1.25-1.23 (t, 6H, CH- (CH 3 ) 2 );
[0105]
P 31 NMR (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ3.56.
[0106]
(3) X is CN
[0107]
Under 5 ℃, the compound (5.20g, 20.0mmol) as shown in Equation 2 in dry THF (30ml) in. T-butyl chloride was added with stirring (1.0M THF solution, 42ml, 42.0mmol). The reaction temperature was raised to 25 ℃, and the mixture was stirred for 30 minutes. After addition of lithium chloride (21.0mmol), was slowly added dropwise a compound of formula 3-2 obtained in Preparation Example 2 (about 28.4 mmol) and THF (30ml) mixed solution, keeping the temperature during at 5 ℃. Bi drops, stirred for 15 hours. With aqueous 1N HCl (25ml) The reaction solution was quenched (HPLC assay Sp: Rp ratio of 6: 1). After further addition of toluene (100ml), temperature was raised to room temperature. The organic layer was washed with 1N HCl, water, 5% Na 2 CO 3 and washed with brine, dried over anhydrous magnesium sulfate, filtered, and the solvent was distilled off under reduced pressure to a solid, was added methylene chloride (20ml), stirred for 5 minutes plus isopropyl ether, stirring was continued for 2 hours, the precipitated solid was filtered off. The solid was dissolved by heating in dichloromethane (60ml), slowly cooled to room temperature and the precipitated crystalline solid. Repeat if necessary obtain pure crystalline sofosbuvir (4.02g, yield 40%, HPLC purity measured 98.8%).
[0108]
1 HNMR (CDCl 3 , 300 MHz): [delta] 8.63 (s, 1H, NH), 7.46 (d, 1H, C6-H), 7.36 (t, 2H, O-aromatic), 7.18-7.24 (m, 3H, m, P-aromatic), 6.20-6.14 (d, 1H, Cl'-H), 5.70-5.68 (d, 1H, C5-H), 5.05-4.97 (m, 1H, CH- (CH 3 ) 2 ) , 4.57-4.41 (m, 2H, C5'-H2), 4.12-4.09 (d, 1H, C3'-H), 4.06-3.79 (m, 3H, C3'-OH, C4'-H, Ala-CH -CH 3 ), 3.79 (s, 1H, Ala-NH), 1.44 (d, 3H, C2'-H3), 1.36-1.34 (d, 3H, Ala-CH 3 ), 1.25-1.23 (t, 6H, CH- (CH 3 ) 2 );
[0109]
P 31 NMR (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ3.56.
//////













